Progeria
| Progeria | |
|---|---|
| Synonyms | Hutchinson–Gilford progeria syndrome (HGPS),[1][2] progeria syndrome[2] |
 | |
| A young girl with progeria (left). A healthy cell nucleus (right, top) and a progeric cell nucleus (right, bottom). | |
| Pronunciation | /proʊˈdʒɪəriə/[3][4] |
| Classification and external resources | |
| Specialty | Medical genetics |
| ICD-10 | E34.8 (ILDS E34.840) |
| ICD-9-CM | 259.8 |
| OMIM | 176670 |
| DiseasesDB | 10704 |
| MedlinePlus | 001657 |
| eMedicine | derm/731 |
| MeSH | D011371 |
| GeneReviews | |
| Orphanet | 740 |
Progeria is an extremely rare genetic disorder in which symptoms resembling aspects of aging are manifested at a very early age.[5] Progeria is one of several progeroid syndromes.[6] The word progeria comes from the Greek words "pro" (πρό), meaning "before" or "premature", and "gēras" (γῆρας), meaning "old age".[7] The disorder has a very low incidence rate, live births.[8] Those born with progeria typically live to their mid teens to early twenties.[9][10] It is a genetic condition that occurs as a new mutation, and is rarely inherited, as carriers usually do not live to reproduce. Although the term progeria applies strictly speaking to all diseases characterized by premature aging symptoms, and is often used as such, it is often applied specifically in reference to Hutchinson–Gilford progeria syndrome (HGPS).
Scientists are particularly interested in progeria because it might reveal clues about the normal process of aging.[11][12][13] Progeria was first described in 1886 by Jonathan Hutchinson.[14] It was also described independently in 1897 by Hastings Gilford.[15] The condition was later named Hutchinson–Gilford progeria syndrome.
Signs and symptoms
Children with progeria usually develop the first symptoms during their first few months of life. The earliest symptoms may include a failure to thrive and a localized scleroderma-like skin condition. As a child ages past infancy, additional conditions become apparent usually around 18–24 months. Limited growth, full-body alopecia (hair loss), and a distinctive appearance (a small face with a shallow recessed jaw, and a pinched nose) are all characteristics of progeria. Signs and symptoms of this progressive disease tend to become more marked as the child ages. Later, the condition causes wrinkled skin, atherosclerosis, kidney failure, loss of eyesight, and cardiovascular problems. Scleroderma, a hardening and tightening of the skin on trunk and extremities of the body, is prevalent. People diagnosed with this disorder usually have small, fragile bodies, like those of elderly people. The face is usually wrinkled, with a larger head in relation to the body, a narrow face and a beak nose. Prominent scalp veins are noticeable (made more obvious by alopecia), as well as prominent eyes. Musculoskeletal degeneration causes loss of body fat and muscle, stiff joints, hip dislocations, and other symptoms generally absent in the non-elderly population. Individuals usually retain typical mental and motor development.
Cause
| Steps in normal cell | Steps in cell with progeria |
|---|---|
| The gene LMNA encodes a protein called prelamin A. | |
| Prelamin A has a farnesyl group attached to its end. | |
| Farnesyl group is removed from prelamin A. | Farnesyl group remains attached to prelamin A. |
| Normal form is called lamin A. | Abnormal form of prelamin A is called progerin. |
| Lamin A is not anchored to the nuclear rim. | Progerin is anchored to the nuclear rim. |
| Normal state of the nucleus. | Abnormally shaped nucleus. |
In normal conditions, the LMNA gene codes for a structural protein called prelamin A which undergoes a series of processing steps before becoming its final form, called lamin A.[16] In one of these steps, after prelamin A is made in the cytoplasm, an enzyme called farnesyl transferase attaches a farnesyl functional group to its carboxyl-terminus. The farnesylated prelamin A is then transported through a nuclear pore to the interior of the nucleus. The farnesyl group allows prelamin A to attach temporarily to the nuclear rim. Once the protein is attached, it is cleaved by a protease, thereby removing the farnesyl group along with a few adjacent amino acids. Failure to remove this farnesyl group permanently affixes the protein to the nuclear rim. After cleavage by the protease, prelamin A is referred to as lamin A. Lamin A, along with lamin B and lamin C, makes up the nuclear lamina, which provides structural support to the nucleus.
Before the late 20th century, research on progeria yielded very little information about the syndrome. In 2003, the cause of progeria was discovered to be a point mutation in position 1824 of the LMNA gene, in which cytosine is replaced with thymine.[17] This mutation creates a 5' cryptic splice site within exon 11, resulting in an abnormally short mature mRNA transcript. This mRNA strand, when translated, yields an abnormal variant of the prelamin A protein whose farnesyl group cannot be removed. Because its farnesyl group cannot be removed, this abnormal protein, referred to as progerin, is permanently affixed to the nuclear rim, and therefore does not become part of the nuclear lamina. Without lamin A, the nuclear lamina is unable to provide the nuclear envelope with adequate structural support, causing it to take on an abnormal shape.[18] Since the support that the nuclear lamina normally provides is necessary for the organizing of chromatin during mitosis, weakening of the nuclear lamina limits the ability of the cell to divide.[19]
To date over 1,400 SNPs of LMNA gene are known.[20] They can manifest in changes on mRNA, splicing or protein (e.g. Arg471Cys,[21] Arg482Gln,[22] Arg527Leu,[23] Arg527Cys,[24] Ala529Val[25]) level.
Progerin may also play a role in normal human aging, since its production is activated in typical senescent cells.[19]
Unlike other "accelerated aging diseases" (such as Werner syndrome, Cockayne syndrome or xeroderma pigmentosum), progeria may not be directly caused by defective DNA repair. Because these diseases cause changes in different aspects of aging, but never in every aspect, they are often called "segmental progerias."[26]
Diagnosis
Diagnosis is suspected according to signs and symptoms, such as skin changes, abnormal growth, and loss of hair. A genetic test for LMNA mutations can confirm the diagnosis of progeria.[27][28]
Treatment
No treatment has proven effective. Most treatment focuses on reducing complications (such as cardiovascular disease) with coronary artery bypass surgery or low-dose aspirin.[29] Children may also benefit from a high-carbohydrate diet.
Growth hormone treatment has been attempted.[30] The use of Morpholinos has also been attempted in order to reduce progerin production. Antisense Morpholino oligonucleotides specifically directed against the mutated exon 11–exon 12 junction in the mutated pre-mRNAs were used.[31]
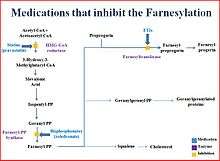
A type of anticancer drug, the farnesyltransferase inhibitors (FTIs), has been proposed, but their use has been mostly limited to animal models.[32] A Phase II clinical trial using the FTI lonafarnib began in May 2007.[33] In studies on the cells another anti-cancer drug, rapamycin, caused removal of progerin from the nuclear membrane through autophagy.[18][34] It has been proved that pravastatin and zoledronate are effective drugs when it comes to the blocking of farnesyl group production.
Farnesyltransferase inhibitors (FTIs) are drugs that inhibit the activity of an enzyme needed in order to make a link between progerin proteins and farnesyl groups. This link generates the permanent attachment of the progerin to the nuclear rim. In progeria, cellular damage can be appreciated because that attachment takes place and the nucleus is not in a normal state. Lonafarnib is an FTI, which means it can avoid this link, so progerin can not remain attached to the nucleus rim and it now has a more normal state.
The delivery of Lonafarnib is not approved by the US Food and Drug Administration (FDA). Therefore, it can only be used in certain clinical trials. Until the treatment of FTIs is implemented in progeria children its effects cannot be known, although its effects on mice seem to be positive.[35]
Pravastatin, traded as Pravachol or Selektine, is included in the family of statins. As well as zoledronate (also known as Zometa and Reclast, which is a bisphosphonate), its utility in HGPS is the prevention of farnesyl group formation, which progerin needs to provoke the disease. Some animal trials have been realized using FTIs or a combination of pravastatin and zoledronate so as to observe whether they are capable of reversing abnormal nuclei.
The results, obtained by blinded electron microscopic analysis and immunofluorescence microscopy, showed that nucleus abnormalities could be reversed in transgenic mice expressing progerin. The reversion was also observed in vivo—cultured cells from human subjects with progeria—due to the action of the pharmacs, which block protein prenylation (transfer of a farnesyl polypeptide to C-terminal cysteine). The authors of that trial add, when it comes to the results, that: "They further suggest that skin biopsy may be useful to determine if protein farnesylation inhibitors are exerting effects in subjects with HGPS in clinical trials".[36]
Unlike FTIs, pravastatin and zoledronate were approved by the U.S. FDA (in 2006 and 2001 respectively), although they are not sold as a treatment for progeria. Pravastatin is used to decrease cholesterol levels and zoledronate to prevent hypercalcaemia.
Rapamycin, also known as Sirolimus, is a macrolide. There are recent studies concerning rapamycin which conclude that it can minimize the phenotypic effects of progeria fibroblasts. Other observed consequences of its use are: abolishment of nuclear blebbing, degradation of progerin in affected cells and reduction of insoluble progerin aggregates formation. All these results do not come from any clinical trial, although it is believed that the treatment might benefit HGPS patients.[18]
A 2012 clinical trial found that the cancer drug Lonafarnib can improve weight gain and other symptoms of progeria.[37]
Prognosis
As there is no known cure, few people with progeria exceed 13 years of age.[38] At least 90% of patients die from complications of atherosclerosis, such as heart attack or stroke.[39]
Mental development is not adversely affected; in fact, intelligence tends to be average to above average.[40] With respect to the features of aging that progeria appears to manifest, the development of symptoms is comparable to aging at a rate eight to ten times faster than normal. With respect to features of aging that progeria does not exhibit, patients show no neurodegeneration or cancer predisposition. They also do not develop the so-called "wear and tear" conditions commonly associated with aging, such as cataracts (caused by UV exposure) and osteoarthritis (caused by mechanical wear).[27]
Although there may not be any successful treatments for progeria itself, there are treatments for the problems it causes, such as arthritic, respiratory, and cardiovascular problems. Sufferers of progeria have normal reproductive development and there are known cases of women with progeria who had delivered healthy offspring.[41]
Epidemiology
A study from the Netherlands has shown an incidence of 1 in 4 million births.[42] Currently, there are 100 known cases in the world. Approximately 140 cases have been reported in medical history.[43] However, the Progeria Research Foundation believes there may be as many as 150 undiagnosed cases worldwide.
Classical Hutchinson–Gilford progeria syndrome is usually caused by a sporadic mutation taking place during the early stages of embryo development. It is almost never passed on from affected parent to child, as affected children rarely live long enough to have children themselves.
There have been only two cases in which a healthy person was known to carry the LMNA mutation that causes progeria. These carriers were identified because they passed it on to their children.[12] One family from India has five children with progeria, though not the classical HGPS type.[44] This family was the subject of a 2005 Bodyshock documentary entitled The 80 Year Old Children. The Vandeweert family of Belgium has two children, Michiel and Amber, with classic HGPS.[45]
Research
Several discoveries have been made that have led to greater understandings and perhaps eventual treatment for this disease.[46][47]
A 2003 report in Nature[48] said that progeria may be a de novo dominant trait. It develops during cell division in a newly conceived zygote or in the gametes of one of the parents. It is caused by mutations in the LMNA (lamin A protein) gene on chromosome 1; the mutated form of lamin A is commonly known as progerin. One of the authors, Leslie Gordon, was a physician who did not know anything about progeria until her own son, Sam, was diagnosed at 22 months. Gordon and her husband, pediatrician Scott Berns, founded the Progeria Research Foundation.[49]
Lamin A
Lamin A is a major component of a protein scaffold on the inner edge of the nucleus called the nuclear lamina that helps organize nuclear processes such as RNA and DNA synthesis.
Prelamin A contains a CAAX box at the C-terminus of the protein (where C is a cysteine and A is any aliphatic amino acids). This ensures that the cysteine is farnesylated and allows prelamin A to bind membranes, specifically the nuclear membrane. After prelamin A has been localized to the cell nuclear membrane, the C-terminal amino acids, including the farnesylated cysteine, are cleaved off by a specific protease. The resulting protein, now lamin A, is no longer membrane-bound, and carries out functions inside the nucleus.
In HGPS, the recognition site that the enzyme requires for cleavage of prelamin A to lamin A is mutated. Lamin A cannot be produced, and prelamin A builds up on the nuclear membrane, causing a characteristic nuclear blebbing.[50] This results in the symptoms of progeria, although the relationship between the misshapen nucleus and the symptoms is not known.
A study that compared HGPS patient cells with the skin cells from young and elderly normal human subjects found similar defects in the HGPS and elderly cells, including down-regulation of certain nuclear proteins, increased DNA damage, and demethylation of histone, leading to reduced heterochromatin.[51] Nematodes over their lifespan show progressive lamin changes comparable to HGPS in all cells but neurons and gametes.[52] These studies suggest that lamin A defects are associated with normal aging.
Mouse model

A mouse model of progeria exists, though in the mouse, the LMNA prelamin A is not mutated. Instead, ZMPSTE24, the specific protease that is required to remove the C-terminus of prelamin A, is missing. Both cases result in the buildup of farnesylated prelamin A on the nuclear membrane and in the characteristic nuclear LMNA blebbing. Fong et al. use a farnesyl transferase inhibitor (FTI) in this mouse model to inhibit protein farnesylation of prelamin A. Treated mice had greater grip strength and lower likelihood of rib fracture and may live longer than untreated mice.[53]
This method does not directly "cure" the underlying cause of progeria. This method prevents prelamin A from going to the nucleus in the first place so that no prelamin A can build up on the nuclear membrane, but equally, there is no production of normal lamin A in the nucleus. Lamin A does not appear to be necessary for life; mice in which the Lmna gene is knocked out show no embryological symptoms (they develop an Emery–Dreifuss muscular dystrophy-like condition postnatally).[54] This implies that it is the buildup of prelamin A in the wrong place, rather than the loss of the normal function of lamin A, that causes the disease.
It was hypothesized that part of the reason that treatment with an FTI such as alendronate is inefficient is due to prenylation by geranylgeranyltransferase. Since statins inhibit geranylgeranyltransferase, the combination of an FTI and statins was tried, and markedly improved "the aging-like phenotypes of mice deficient in the metalloproteinase Zmpste24, including growth retardation, loss of weight, lipodystrophy, hair loss, and bone defects".[55]
DNA repair
Repair of DNA double-strand breaks can occur by either of two processes, non-homologous end joining (NHEJ) or homologous recombination (HR). A-type lamins promote genetic stability by maintaining levels of proteins that have key roles in NHEJ and HR.[56] Mouse cells deficient for maturation of prelamin A show increased DNA damage and chromosome aberrations and have increased sensitivity to DNA damaging agents.[57] In progeria, the inability to adequately repair DNA damages due to defective A-type lamin may cause aspects of premature aging[58] (also see DNA damage theory of aging).
Popular culture
Perhaps one of the earliest influences of progeria on popular culture occurred in the 1922 short story The Curious Case of Benjamin Button by F. Scott Fitzgerald (and later released as a feature film in 2008). The main character, Benjamin Button, is born as a seventy-year-old man and ages backwards; it has been suggested that this was inspired by progeria.[59]
Charles Dickens may have described a case of progeria in the Smallweed family of Bleak House, specifically in the grandfather and his grandchildren, Judy and twin brother Bart.[60]
A 2009 Bollywood movie, Paa, was made about the condition; in it, the lead (Amitabh Bachchan) played a 12-year-old child affected by progeria.
In the 1983 film The Hunger, progeria was the focus of study by Susan Sarandon's character, Dr. Sarah Roberts.
The 1984 film The Three Wishes of Billy Grier stars Ralph Macchio as a teenager who tries to fulfill his wishes before he dies from the disease.
In 1987, 12 year old Mickey Hays, who had progeria, appeared along with Jack Elam in the documentary I Am Not a Freak.[61] Elam and Hays first met during the filming of the 1986 film The Aurora Encounter.,[62] in which Hays was cast as an alien. The friendship that developed lasted until Hays passed in 1992, age 20. Elam said, "You know I've met a lot of people, but I've never met anybody that got next to me like Mickey."
The 1996 movie Jack deals with the eponymous character (Robin Williams) who has a genetic disorder similar to progeria and the difficulties he faces fitting into society.
The 2006 movie Renaissance deals with progeria.
"Young at Heart", the sixteenth episode of the first season of the television show The X-Files, features a violent criminal who has seemingly grown younger due to treatment by a corrupt doctor, who had developed his technique by experimenting on progeria sufferers.
In Tad Williams' novel series Otherland, one of the main characters suffers from progeria.
Harold Kushner's 1978 book When Bad Things Happen to Good People, which explores God and the problem of evil, was written in response to his 14-year-old son's death due to progeria.
South African artist/hip hop artist Leon Botha was one of the oldest known progeria sufferers, surviving to the age of 26 before his death in June 2011.
In Chuck Palahniuk's 2005 novel Haunted the main villain is Mr. Whittier, a 13 year old sufferer of progeria. Mr. Whittier tricked middle-aged married women to sleep with him by telling them that he was an eighteen-year-old virgin, he then blackmailed them into giving him money by telling them that he would charge them with statutory rape if they did not.
Meg Casey, a Milford, CT artist and spokesperson for the handicapped, was born October 1, 1955 and died May 26, 1985. She survived for 29 years with progeria.[63][64][65]
In the Star Trek: Voyager episode "Scientific Method", Chakotay begins to rapidly age as a result of an alien science experiment. Progeria is posited as one possible cause to the rapid aging, but The Doctor notes that there had never been an adult case, and that the disorder had been eradicated sometime during the 22nd century, approximately 200 years before the events of that episode.
Life According to Sam was a 2013 documentary on Foxborough High School (Foxborough, Massachusetts) student Sam Berns. He was age 17 when he died of the disease, January 10, 2014, and a fan of the New England Patriots. Had he lived another day, he would have served as the team's honorary captain in their playoff game versus the Indianapolis Colts.[66] Produced by Sean Fine and Andrea Nix, the film explains progeria and follows the process of finding a cure for it.[67] In an interview, Berns had said that the most important thing people should know about him is that he had a very happy life.[68]
My Brilliant Life was a 2014 South Korean film about two seventeen-year-olds who become parents to Ah-reum, who was born and was diagnosed with progeria.
See also
- Biogerontology
- Sam Berns, a Massachusetts boy who suffered from progeria and helped raise awareness of the disease.
- Degenerative disease
- Hayley Okines, an English girl who had progeria and was known for spreading awareness
- Laminopathies
- Lipodystrophy
- Lizzie Velásquez, an American motivational speaker whose medical condition approximates neonatal progeroid syndrome
References
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 574. ISBN 0-7216-2921-0.
- 1 2 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- ↑ Dictionary Reference: Progeria
- ↑ The Free Dictionary: Progeria
- ↑ Sinha, Jitendra Kumar; Ghosh, Shampa; Raghunath, Manchala (May 2014). "Progeria: a rare genetic premature ageing disorder". Indian J Med Res. 139 (5): 667–74. PMC 4140030
 . PMID 25027075.
. PMID 25027075. - ↑ Ramírez CL, Cadiñanos J, Varela I, Freije JM, López-Otín C (2007). "Human progeroid syndromes, aging and cancer: new genetic and epigenetic insights into old questions". Cell. Mol. Life Sci. 64: 155–70. doi:10.1007/s00018-006-6349-3. PMID 17131053.
- ↑ http://lexbook.net/en/progeria
- ↑ Progeria, Incidence of Progeria and HGPS.
- ↑ Ewell Steve Roach; Van S. Miller (2004). Neurocutaneous Disorders. Cambridge University Press. p. 150. ISBN 978-0-521-78153-4.
- ↑ Kwang-Jen Hsiao (1998). Advances in Clinical Chemistry:33. Academic Press. p. 10. ISBN 0-12-010333-8.
- ↑ McClintock D, Ratner D, Lokuge M; et al. (2007). Lewin, Alfred, ed. "The Mutant Form of Lamin A that Causes Hutchinson–Gilford Progeria Is a Biomarker of Cellular Aging in Human Skin". PLoS ONE. 2 (12): e1269. Bibcode:2007PLoSO...2.1269M. doi:10.1371/journal.pone.0001269. PMC 2092390. PMID 18060063.
- 1 2 Korf B (2008). "Hutchinson–Gilford progeria syndrome, aging, and the nuclear lamina". N. Engl. J. Med. 358 (6): 552–5. doi:10.1056/NEJMp0800071. PMID 18256390.
- ↑ Merideth MA, Gordon LB, Clauss S, et al. (2008). "Phenotype and course of Hutchinson–Gilford progeria syndrome". N. Engl. J. Med. 358 (6): 592–604. doi:10.1056/NEJMoa0706898. PMC 2940940. PMID 18256394.
- ↑ Hutchinson J (1886). "Case of congenital absence of hair, with atrophic condition of the skin and its appendages, in a boy whose mother had been almost wholly bald from alopecia areata from the age of six". Lancet. I (3272): 923. doi:10.1016/S0140-6736(02)06582-0.
- ↑ Gilford H; Shepherd, RC (1904). "Ateleiosis and progeria: continuous youth and premature old age". Brit. Med. J. 2 (5157): 914–8. PMC 1990667. PMID 14409225.
- ↑ LMNA At Genes At Genetics Home Reference
- ↑ De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, CL.; et al. (Jun 2003). "Lamin a truncation in Hutchinson–Gilford progeria". Science. 300 (5628): 2055. doi:10.1126/science.1084125. PMID 12702809.
- 1 2 3 Cao, K.; Collins, F. S. (June 2011). "Rapamycin Reverses Cellular Phenotypes and Enhances Mutant Protein Clearance in Hutchinson–Gilford Progeria Syndrome Cells". Science Translational Medicine. 3 (89): 89ra58. doi:10.1126/scitranslmed.3002346. PMID 21715679.
- 1 2 Norris, J. (2011-10-21). "Aging Disease in Children Sheds Light on Normal Aging". UCSF web site. UCSF. Retrieved 2011-10-25.
- ↑ "LMNA Gene". GeneCards. Retrieved June 6, 2015.
- ↑ Zirn B, Kress W, Grimm T, Berthold LD; et al. (2008). "Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy". Am J Med Genet A. 146A (8): 1049–1054. doi:10.1002/ajmg.a.32259. PMID 18348272.
- ↑ Cao H, Hegele RA; Hegele (2002). "Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy". Hum. Molec. Genet. 9 (1): 109–12. doi:10.1093/hmg/9.1.109. PMID 10587585.
- ↑ Al-Haggar M, Madej-Pilarczyk A, Kozlowski L, Bujnicki JM, Yahia S, Abdel-Hadi D, Shams A, Ahmad N, Hamed S, Puzianowska-Kuznicka M; Madej-Pilarczyk; Kozlowski; Bujnicki; Yahia; Abdel-Hadi; Shams; Ahmad; Hamed; Puzianowska-Kuznicka (2012). "A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome". Eur J Hum Genet. 20 (11): 1134–40. doi:10.1038/ejhg.2012.77. PMC 3476705. PMID 22549407.
- ↑ Agarwal AK, Kazachkova I, Ten S, Garg A; Kazachkova; Ten; Garg (2008). "Severe mandibuloacral dysplasia-associated lipodystrophy and progeria in a young girl with a novel homozygous Arg527Cys LMNA mutation". J Clin Endocrinol Metab. 93 (12): 4617–4623. doi:10.1210/jc.2008-0123. PMC 2626450. PMID 18796515.
- ↑ Garg A, Cogulu O, Ozkinay F, Onay H, Agarwal AK; Cogulu; Ozkinay; Onay; Agarwal (2005). "A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia". J. Clin. Endocrinol. Metab. 90 (9): 5259–64. doi:10.1210/jc.2004-2560. PMID 15998779.
- ↑ Best,BP (2009). "Nuclear DNA damage as a direct cause of aging" (PDF). Rejuvenation Research. 12 (3): 199–208. doi:10.1089/rej.2009.0847. PMID 19594328.
- 1 2 "Learning About Progeria". genome.gov. Retrieved 2008-03-17.
- ↑ "Progeria Research Foundation | The PRF Diagnostic Testing Program". Retrieved 16 November 2011.
- ↑ "Progeria: Treatment". MayoClinic.com. Retrieved 2008-03-17.
- ↑ Sadeghi-Nejad A, Demmer L; Demmer (2007). "Growth hormone therapy in progeria". J. Pediatr. Endocrinol. Metab. 20 (5): 633–7. doi:10.1515/JPEM.2007.20.5.633. PMID 17642424.
- ↑ Scaffidi, P., Misteli, T.; Misteli (2005). "Reversal of the cellular phenotype in the premature aging disease Hutchinson–Gilford progeria syndrome". Nat. Med. 11 (4): 440–5. doi:10.1038/nm1204. PMC 1351119. PMID 15750600.
- ↑ Meta M, Yang SH, Bergo MO, Fong LG, Young SG; Yang; Bergo; Fong; Young (2006). "Protein farnesyltransferase inhibitors and progeria". Trends Mol Med. 12 (10): 480–7. doi:10.1016/j.molmed.2006.08.006. PMID 16942914.
- ↑ Clinical trial number NCT00425607 for "Phase II Trial of Lonafarnib (a Farnesyltransferase Inhibitor) for Progeria" at ClinicalTrials.gov
- ↑ Staff writer (2011). "New Drug Hope for 'Aging' Kids". Nature. 333 (6039): 142. doi:10.1126/science.333.6039.142-b.
- ↑ Capell BC; et al. (2005). "Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson–Gilford progeria syndrome". Proc Natl Acad Sci USA. 102 (36): 12879–84. Bibcode:2005PNAS..10212879C. doi:10.1073/pnas.0506001102. PMC 1200293. PMID 16129833.
- ↑ Wang Y; et al. (September–October 2010). "Blocking protein farnesylation improves nuclear shape abnormalities in keratinocytes of mice expressing the prelamin A variant in Hutchinson–Gilford progeria syndrome". Landes Bioscience. 1 (5): 432–9. doi:10.4161/nucl.1.5.12972. PMC 3037539. PMID 21326826.
- ↑ Hamilton, Jon (September 22, 2012). "Experimental Drug Is First To Help Kids With Premature-Aging Disease". NPR. Retrieved 21 October 2012.
- ↑ Steve Sternberg (April 16, 2003). "Gene found for rapid aging disease in children". USA Today. Retrieved 2006-12-13.
- ↑ "Progeria". MayoClinic.com. Retrieved 2008-03-17.
- ↑ Brown WT (June 1992). "Progeria: a human-disease model of accelerated aging". Am. J. Clin. Nutr. 55 (6 Suppl): 1222S–4S. PMID 1590260.
- ↑ Corcoy R, Aris A, de Leiva A (1989). "Fertility in a case of progeria". Am. J. Med. Sci. 297: 383–4. doi:10.1097/00000441-198906000-00010. PMID 2735343.
- ↑ Hennekam RC (2006). "Hutchinson–Gilford progeria syndrome: review of the phenotype". Am. J. Med. Genet. A. 140 (23): 2603–24. doi:10.1002/ajmg.a.31346. PMID 16838330.
- ↑ "Progeria Info". Retrieved 2013-11-28.
- ↑ Grant, Matthew (22 February 2005). "Family tormented by ageing disease". BBC News. Retrieved on 3 May 2009.
- ↑ Hope, Alan (3 June 2009). "Face of Flanders: Michiel Vandeweert". Flanders Today. Retrieved on 3 September 2009.
- ↑ Capell BC, Collins FS, Nabel EG; Collins; Nabel (2007). "Mechanisms of cardiovascular disease in accelerated aging syndromes". Circ. Res. 101 (1): 13–26. doi:10.1161/CIRCRESAHA.107.153692. PMID 17615378.
- ↑ Gordon, Leslie B.; Cao, Kan; Collins, Francis S. (2012). "Progeria: Translational insights from cell biology". J Cell Biol. 199 (1): 9–13. doi:10.1083/jcb.201207072. PMID 23027899.
- ↑ M. Eriksson; et al. (2003). "Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome" (PDF). Nature. 423 (6937): 293–298. Bibcode:2003Natur.423..293E. doi:10.1038/nature01629. PMID 12714972.
- ↑ "Family Crisis Becomes Scientific Quest", Science, 300(5621), 9 May 2003.
- ↑ Lans H, Hoeijmakers JH; Hoeijmakers (2006). "Cell biology: ageing nucleus gets out of shape". Nature. 440 (7080): 32–4. Bibcode:2006Natur.440...32L. doi:10.1038/440032a. PMID 16511477.
- ↑ Scaffidi P, Misteli T; Misteli (May 19, 2006). "Lamin A-dependent nuclear defects in human aging". Science. 312 (5776): 1059–63. Bibcode:2006Sci...312.1059S. doi:10.1126/science.1127168. PMC 1855250. PMID 16645051.
- ↑ Haithcock E, Dayani Y, Neufeld E; et al. (2005). "Age-related changes of nuclear architecture in Caenorhabditis elegans". Proc. Natl. Acad. Sci. U.S.A. 102 (46): 16690–5. Bibcode:2005PNAS..10216690H. doi:10.1073/pnas.0506955102. PMC 1283819. PMID 16269543.
- ↑ Fong, L. G.; et al. (March 17, 2006). "A Protein Farnesyltransferase Inhibitor Ameliorates Disease in a Mouse Model of Progeria". Science. 311 (5767): 1621–3. Bibcode:2006Sci...311.1621F. doi:10.1126/science.1124875. PMID 16484451.
- ↑ Sullivan; et al. (November 29, 1999). "Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy". J Cell Biol. 147 (5): 913–20. doi:10.1083/jcb.147.5.913. PMC 2169344. PMID 10579712.
- ↑ Varela I, Pereira S, Ugalde AP, et al. (2008). "Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging". Nat. Med. 14 (7): 767–72. doi:10.1038/nm1786. PMID 18587406.
- ↑ Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, Stewart CL, Zhang J, Gonzalo S (2011). "A dual role for A-type lamins in DNA double-strand break repair". Cell Cycle. 10 (15): 2549–60. doi:10.4161/cc.10.15.16531. PMC 3180193. PMID 21701264.
- ↑ Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadiñanos J, López-Otín C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z (2005). "Genomic instability in laminopathy-based premature aging". Nat. Med. 11 (7): 780–5. doi:10.1038/nm1266. PMID 15980864.
- ↑ Bernstein H, Payne CM, Bernstein C, Garewal H, Dvorak K (2008). Cancer and aging as consequences of un-repaired DNA damage. In: New Research on DNA Damages (Editors: Honoka Kimura and Aoi Suzuki) Nova Science Publishers, Inc., New York, Chapter 1, pp. 1-47. open access, but read only https://www.novapublishers.com/catalog/product_info.php?products_id=43247 ISBN 978-1604565812
- ↑ Maloney WJ (October 2009). "Hutchinson–Gilford Progeria Syndrome: Its Presentation in F. Scott Fitzgerald's Short Story 'The Curious Case of Benjamin Button' and its Oral Manifestations". J. Dent. Res. 88 (10): 873–6. doi:10.1177/0022034509348765. PMID 19783794.
- ↑ Singh V (2010). "Reflections: neurology and the humanities. Description of a family with progeria by Charles Dickens". Neurology. 75 (6): 571. doi:10.1212/WNL.0b013e3181ec7f6c. PMID 20697111.
- ↑ "I Am Not a Freak" (1987) at the Internet Movie Database. Retrieved 2009-11-27.
- ↑ "The Aurora Encounter" (1986) at the Internet Movie Database. Retrieved 2009-11-27.
- ↑ BURT A. FOLKART (May 30, 1985). "Oldest Survivor of Rare Aging Disease : Meg Casey, 29, Dies After an Eloquent Struggle". Los Angeles Times. Retrieved July 18, 2016.
- ↑ United Press International (May 28, 1985). "Oldest Progeria Survivor Dies At 29 In Connecticut". Orlando Sentinel. Retrieved July 18, 2016.
- ↑ Robin Marantz Henig (January 30, 2005). "Racing with Sam". The New York Times. Retrieved July 18, 2016.
- ↑ Greg Botelho (12 January 2014). "Beloved teen Sam Berns dies at 17 after suffering from rare disease". CNN.
- ↑ Life According to Sam. HBO Documentaries.
- ↑ Berns, Sam (December 13, 2013). "My philosophy for a happy life". Ted Talks. Retrieved June 6, 2015 – via YouTube.
External links
| Wikimedia Commons has media related to progeria. |
- Progeria Research Foundation
- Progeria News and Media Collection
- GeneReview/NIH/UW entry on Hutchinson–Gilford progeria syndrome
- Segmental progeria
- "ABC 20/20 special news program about Progeria, with Barbara Walters"