Pituitary apoplexy
| Pituitary apoplexy | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E23.6 |
| ICD-9-CM | 253.8 |
| DiseasesDB | 10049 |
| MedlinePlus | 001167 |
| eMedicine | oph/471 |
| MeSH | D010899 |
Pituitary apoplexy or pituitary tumor apoplexy is bleeding into or impaired blood supply of the pituitary gland at the base of the brain. This usually occurs in the presence of a tumor of the pituitary, although in 80% of cases this has not been diagnosed previously. The most common initial symptom is a sudden headache, often associated with a rapidly worsening visual field defect or double vision caused by compression of nerves surrounding the gland. This is followed in many cases by acute symptoms caused by lack of secretion of essential hormones, predominantly adrenal insufficiency.[1]
The diagnosis is achieved with magnetic resonance imaging and blood tests. Treatment is by the timely correction of hormone deficiencies, and in many cases surgical decompression is required. Many people who have had a pituitary apoplexy develop pituitary hormone deficiencies and require long-term hormone supplementation. The first case of the disease was recorded in 1898.[1]
Signs and symptoms
Acute symptoms
The initial symptoms of pituitary apoplexy are related to the increased pressure in and around the pituitary gland. The most common symptom, in over 95% of cases, is a sudden-onset headache located behind the eyes or around the temples. It is often associated with nausea and vomiting.[1][2][3] Occasionally, the presence of blood leads to irritation of the lining of the brain, which may cause neck rigidity and intolerance to bright light, as well as a decreased level of consciousness.[1][2][3] This occurs in 24% of cases.[4]

Pressure on the part of the optic nerve known as the chiasm, which is located above the gland, leads to loss of vision on the outer side of the visual field on both sides, as this corresponds to areas on the retinas supplied by these parts of the optic nerve; it is encountered in 75% of cases.[1] Visual acuity is reduced in half, and over 60% have a visual field defect.[2][4] The visual loss depends on which part of the nerve is affected. If the part of the nerve between the eye and the chiasm is compressed, the result is vision loss in one eye. If the part after the chiasm is affected, visual loss on one side of the visual field occurs.[2]
Adjacent to the pituitary lies a part of the skull base known as the cavernous sinus. This contains a number of nerves that control the eye muscles. 70% of people with pituitary apoplexy experience double vision due to compression of one of the nerves. In half of these cases, the oculomotor nerve (the third cranial nerve), which controls a number of eye muscles, is affected. This leads to diagonal double vision and a dilated pupil. The fourth (trochlear) and sixth (abducens) cranial nerves are located in the same compartment and can cause diagonal or horizontal double vision, respectively.[1] The oculomotor nerve is predominantly affected as it lies closest to the pituitary.[2][5] The cavernous sinus also contains the carotid artery, which supplies blood to the brain; occasionally, compression of the artery can lead to one-sided weakness and other symptoms of stroke.[1][2][4]
Endocrine dysfunction
The pituitary gland consists of two parts, the anterior (front) and posterior (back) pituitary. Both parts release hormones that control numerous other organs. In pituitary apoplexy, the main initial problem is a lack of secretion of adrenocorticotropic hormone (ACTH, corticotropin), which stimulates the secretion of cortisol by the adrenal gland. This occurs in 70% of those with pituitary apoplexy. A sudden lack of cortisol in the body leads to a constellation of symptoms called "adrenal crisis" or "Addisonian crisis" (after a complication of Addison's disease, the main cause of adrenal dysfunction and low cortisol levels).[1] The main problems are low blood pressure (particularly on standing), low blood sugars (which can lead to coma) and abdominal pain; the low blood pressure can be life-threatening and requires immediate medical attention.[6]
Hyponatremia, an unusually low level of sodium in the blood that may cause confusion and seizures, is found in 40% of cases. This may be caused by low cortisol levels or by inappropriate release of antidiuretic hormone (ADH) from the posterior pituitary.[1] Several other hormonal deficiencies may develop in the subacute phase. 50% have a deficiency in thyroid-stimulating hormone (TSH), leading to undersecretion of thyroid hormone by the thyroid gland and characteristic symptoms such as fatigue, weight gain, and cold intolerance. 75% develop a deficiency to gonadotropins (LH and FSH), which control the reproductive hormone glands. This leads to a disrupted menstrual cycle, infertility and decreased libido.[1]
Causes
Almost all cases of pituitary apoplexy arise from a pituitary adenoma, a benign tumor of the pituitary gland. In 80%, the patient has been previously unaware of this (although some will retrospectively report associated symptoms).[1] It was previously thought that particular types of pituitary tumors were more prone to apoplexy than others, but this has not been confirmed.[2] In absolute terms, only a very small proportion of pituitary tumors eventually undergoes apoplexy. In an analysis of incidentally found pituitary tumors, apoplexy occurred in 0.2% annually, but the risk was higher in tumors larger than 10 mm ("macroadenomas") and tumors that were growing more rapidly; in a meta-analysis, not all these associations achieved statistical significance.[7]
The majority of cases (60–80%) are not precipitated by a particular cause.[2] A quarter has a history of high blood pressure,[1] but this is a common problem in the general population, and it is not clear whether it significantly increase the risk of apoplexy.[8] A number of cases has been reported in association with particular conditions and situations; it is uncertain whether these were in fact causative.[8] Amongst reported associations are surgery (especially coronary artery bypass graft, where there are significant fluctuations in the blood pressure), disturbances in blood coagulation or medication that inhibits coagulation, radiation therapy to the pituitary, traumatic brain injury, pregnancy (during which the pituitary enlarges) and treatment with estrogens. Hormonal stimulation tests of the pituitary have been reported to provoke episodes. Treatment of prolactinomas (pituitary adenomas that secrete prolactin) with dopamine agonist drugs, as well as withdrawal of such treatment, has been reported to precipitate apoplexy.[1][2][4]
Hemorrhage from a Rathke's cleft cyst, a remnant of Rathke's pouch that normally regresses after embryological development, may cause symptoms that are indistinguishable from pituitary apoplexy.[4] Pituitary apoplexy is regarded by some as distinct from Sheehan's syndrome, where the pituitary undergoes infarction as a result of prolonged very low blood pressure, particularly when caused by bleeding after childbirth. This condition usually occurs in the absence of a tumor.[4] Others regard Sheehan's syndrome as a form of pituitary apoplexy.[3][9]
Mechanism
The pituitary gland is located in a recess in the skull base known as the sella turcica ("Turkish saddle", after its shape). It is attached to the hypothalamus, a part of the brain, by a stalk that also contains the blood vessels that supply the gland. It is unclear why pituitary tumors are five times more likely to bleed than other tumors in the brain. There are various proposed mechanisms by which a tumor can increase the risk of either infarction (insufficient blood supply leading to tissue dysfunction) or hemorrhage.[2] The pituitary gland normally derives its blood supply from vessels that pass through the hypothalamus, but tumors develop a blood supply from the nearby inferior hypophyseal artery that generates a higher blood pressure, possibly accounting for the risk of bleeding. Tumors may also be more sensitive to fluctuations in blood pressure, and the blood vessels may show structural abnormalities that make them vulnerable to damage. It has been suggested that infarction alone causes milder symptoms than either hemorrhage or hemorrhagic infarction (infarction followed by hemorrhage into the damaged tissue).[4] Larger tumors are more prone to bleeding, and more rapidly growing lesions (as evidenced by detection of increased levels of the protein PCNA) may also be at a higher risk of apoplexy.[2]
After an apoplexy, the pressure inside the sella turcica rises, and surrounding structures such as the optic nerve and the contents of the cavernous sinus are compressed. The raised pressure further impairs the blood supply to the pituitary hormone-producing tissue, leading to tissue death due to insufficient blood supply.[2]
Diagnosis
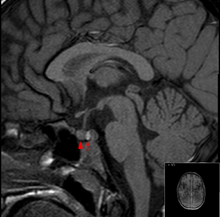
It is recommended that magnetic resonance imaging (MRI) scan of the pituitary gland is performed if the diagnosis is suspected; this has a sensitivity of over 90% for detecting pituitary apoplexy; it may demonstrate infarction (tissue damage due to a decreased blood supply) or hemorrhage.[1] Different MRI sequences can be used to establish when the apoplexy occurred, and the predominant form of damage (hemorrhage or infarction).[2] If MRI is not suitable (e.g. due to claustrophobia or the presence of metal-containing implants), a computed tomography (CT) scan may demonstrate abnormalities in the pituitary gland, although it is less reliable.[1] Many pituitary tumors (25%) are found to have areas of hemorrhagic infarction on MRI scans, but apoplexy is not said to exist unless it is accompanied by symptoms.[1][4]
In some instances, lumbar puncture may be required if there is a suspicion that the symptoms might be caused by other problems (meningitis or subarachnoid hemorrhage). This is the examination of the cerebrospinal fluid that envelops the brain and the spinal cord; the sample is obtained with a needle that is passed under local anesthetic into the spine. In pituitary apoplexy the results are typically normal, although abnormalities may be detected if blood from the pituitary has entered the subarachnoid space.[2][4] If there is remaining doubt about the possibility of subarachnoid hemorrhage (SAH), a magnetic resonance angiogram (MRI with a contrast agent) may be required to identify aneurysms of the brain blood vessels, the most common cause of SAH.[10]
Professional guidelines recommend that if pituitary apoplexy is suspected or confirmed, the minimal blood tests performed should include a complete blood count, urea (a measure of renal function, usually performed together with creatinine), electrolytes (sodium and potassium), liver function tests, routine coagulation testing, and a hormonal panel including IGF-1, growth hormone, prolactin, luteinizing hormone, follicle-stimulating hormone, thyroid-stimulating hormone, thyroid hormone, and either testosterone in men or estradiol in women.[1]
Visual field testing is recommended as soon as possible after diagnosis,[1][4] as it quantifies the severity of any optic nerve involvement, and may be required to decide on surgical treatment.[1]
Treatment
The first priority in suspected or confirmed pituitary apoplexy is stabilization of the circulatory system. Cortisol deficiency can cause severe low blood pressure.[1][6] Depending on the severity of the illness, admission to a high dependency unit (HDU) may be required.[1]
Treatment for acute adrenal insufficiency requires the administration of intravenous saline or dextrose solution; volumes of over two liters may be required in an adult.[6] This is followed by the administration of hydrocortisone, which is pharmaceutical grade cortisol, intravenously or into a muscle.[4][6] The drug dexamethasone has similar properties,[6] but its use is not recommended unless it is required to reduce swelling in the brain around the area of hemorrhage.[1] Some are well enough not to require immediate cortisol replacement; in this case, blood levels of cortisol are determined at 9:00 AM (as cortisol levels vary over the day). A level below 550 nmol/l indicates a need for replacement.[1]
The decision on whether to surgically decompress the pituitary gland is complex and mainly dependent on the severity of visual loss and visual field defects. If visual acuity is severely reduced, there are large or worsening visual field defects, or the level of consciousness falls consistently, professional guidelines recommend that surgery is performed.[1] Most commonly, operations on the pituitary gland are performed through transsphenoidal surgery. In this procedure, surgical instruments are passed through the nose towards the sphenoid bone, which is opened to give access to the cavity that contains the pituitary gland.[1][4] Surgery is most likely to improve vision if there was some remaining vision before surgery,[4] and if surgery is undertaken within a week of the onset of symptoms.[4][11]
Those with relatively mild visual field loss or double vision only may be managed conservatively, with close observation of the level of consciousness, visual fields, and results of routine blood tests. If there is any deterioration, or expected spontaneous improvement does not occur, surgical intervention may still be indicated.[1][4] If the apoplexy occurred in a prolactin-secreting tumor, this may respond to dopamine agonist treatment.[4][11]
After recovery, people who have had pituitary apoplexy require follow-up by an endocrinologist to monitor for long-term consequences. MRI scans are performed 3–6 months after the initial episode and subsequently on an annual basis.[1] If after surgery some tumor tissue remains, this may respond to medication, further surgery, or radiation therapy with a "gamma knife".[4]
Prognosis
In larger case series, the mortality was 1.6% overall. In the group of patients who were unwell enough to require surgery, the mortality was 1.9%, with no deaths in those who could be treated conservatively.[8]
After an episode of pituitary apoplexy, 80% of people develop hypopituitarism and require some form of hormone replacement therapy.[1][2] The most common problem is growth hormone deficiency, which is often left untreated[1][4] but may cause decreased muscle mass and strength, obesity and fatigue.[12] 60–80% require hydrocortisone replacement (either permanently or when unwell), 50–60% need thyroid hormone replacement, and 60–80% of men require testosterone supplements. Finally, 10–25% develop diabetes insipidus, the inability to retain fluid in the kidneys due to a lack of the pituitary antidiuretic hormone. This may be treated with the drug desmopressin,[1] which can be applied as a nose spray or taken by mouth.[13]
Epidemiology
Pituitary apoplexy is rare.[1][2] Even in people with a known pituitary tumor, only 0.6–10% experience apoplexy; the risk is higher in larger tumors.[2] Based on extrapolations from existing data, one would expect 18 cases of pituitary apoplexy per one million people every year; the actual figure is probably lower.[14]
The average age at onset is 50; cases have reported in people between 15 and 90 years old.[14] Men are affected more commonly than women,[2] with a male-to-female ratio of 1.6.[4] The majority of the underlying tumors are "null cell" or nonsecretory tumors, which do not produce excessive amounts of hormones; this might explain why the tumor has often gone undetected prior to an episode of apoplexy.[4]
History
The first case description of pituitary apoplexy has been attributed to the American neurologist Pearce Bailey in 1898.[15][16] This was followed in 1905 by a further report from the German physician Bleibtreu.[4][17] Surgery for pituitary apoplexy was described in 1925.[16][18] Before the introduction of steroid replacement, the mortality from pituitary apoplexy approximated 50%.[4]
The name of the condition was coined in 1950 in a case series by physicians from Boston City Hospital and Harvard Medical School.[1][19] The term "apoplexy" was applied as it referred to both necrosis and bleeding into pituitary tumors.[19]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 Rajasekaran S, Vanderpump M, Baldeweg S, et al. (Jan 2011). "UK guidelines for the management of pituitary apoplexy". Clin Endocrinol (Oxf). 74 (1): 9–20. doi:10.1111/j.1365-2265.2010.03913.x. PMID 21044119.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Murad-Kejbou S, Eggenberger E (Nov 2009). "Pituitary apoplexy: evaluation, management, and prognosis". Current Opinion in Ophthalmology. 20 (6): 456–61. doi:10.1097/ICU.0b013e3283319061. PMID 19809320.
- 1 2 3 Melmed S, Jameson JL (2005). "Disorders of the anterior pituitary and hypothalamus". In Kasper DL, Braunwald E, Fauci AS, et al. Harrison's Principles of Internal Medicine (16th ed.). New York, NY: McGraw-Hill. pp. 2076–97. ISBN 0-07-139140-1.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 Nawar RN, AbdelMannan D, Selman WR, Arafah BM (Mar–Apr 2008). "Pituitary tumor apoplexy: a review". J. Intens. Care Med. 23 (2): 75–90. doi:10.1177/0885066607312992. PMID 18372348.
- ↑ Bruce BB, Biousse V, Newman NJ (Jul 2007). "Third nerve palsies". Semin Neurol. 27 (3): 257–68. doi:10.1055/s-2007-979681. PMID 17577867.
- 1 2 3 4 5 de Herder WW, van der Lely AJ (May 2003). "Addisonian crisis and relative adrenal failure". Rev Endocr Metab Disord. 4 (2): 143–7. doi:10.1023/A:1022938019091. PMID 12766542.
- ↑ Fernández-Balsells MM, Murad MH, Barwise A, et al. (April 2011). "Natural history of nonfunctioning pituitary adenomas and incidentalomas: a systematic review and metaanalysis". J. Clin. Endocrinol. Metab. 96 (4): 905–12. doi:10.1210/jc.2010-1054. PMID 21474687.
- 1 2 3 Russell SJ, Miller K (2008). "Pituitary apoplexy". In Swearingen B, Biller BM. Diagnosis and management of pituitary disorders. Totowa, NJ: Humana Press. p. 368. ISBN 1-58829-922-8.
- ↑ Thorner MO, Vance ML, Horvath E, Kovacs K (1992). "The anterior pituitary". In Wilson JD, Foster DW. Williams Textbook of Endocrinology, 8th edition. Philadelphia: W.B. Saunders. pp. 294–5. ISBN 0-7216-9514-0.
- ↑ Post KD, Shiau JS, Walsh J (2008). "Pituitary apoplexy". In Loftus CM. Neurosurgical Emergencies (2nd ed.). New York, NY: Thieme Publishing Group. pp. 78–83. ISBN 3-13-135052-0.
- 1 2 Adams CB (2003). "The surgery of pituitary tumours". In Wass JA, Shalet SM. Oxford Textbook of Endocrinology and Diabetes. Oxford: Oxford University Press. pp. 161–2. ISBN 0-19-263045-8.
- ↑ van Aken MO, Lamberts SW (2005). "Diagnosis and treatment of hypopituitarism: an update". Pituitary. 8 (3-4): 183–91. doi:10.1007/s11102-006-6039-z. PMID 16508719.
- ↑ Jane JA Jr; Vance ML; Laws ER (2006). "Neurogenic diabetes insipidus". Pituitary. 9 (4): 327–9. doi:10.1007/s11102-006-0414-7. PMID 17080264.
- 1 2 Russell SJ, Miller K (2008). "Pituitary apoplexy". In Swearingen B, Biller BM. Diagnosis and management of pituitary disorders. Totowa, NJ: Humana Press. p. 356. ISBN 1-58829-922-8.
- ↑ Bailey P (1898). "Pathological report of a case of akromegaly, with special reference to the lesions in the hypophysis cerebri and in the thyroid gland; and a case of haemorrhage into the pituitary". Phila Med J. 1: 789–92.
- 1 2 Russell SJ, Klahr Miller K (2008). "Pituitary apoplexy". In Swearingen B, Biller BM. Diagnosis and management of pituitary disorders. Totowa, NJ: Humana Press. p. 354. ISBN 1-58829-922-8.
- ↑ Bleibtreu L (1905). "Ein Fall von Akromegalia (Zerstorung der Hypophysis durch Blutung)". Munch Med Wochenschr (in German). 41: 2079–80.
- ↑ Dott NM, Bailey P, Cushing H (1925). "A consideration of the hypophyseal adenomata". Br J Surg. 13: 314–66. doi:10.1002/bjs.1800135009.
- 1 2 Brougham M, Heusner AP, Adams RD (Sep 1950). "Acute degenerative changes in adenomas of the pituitary body--with special reference to pituitary apoplexy". J Neurosurg. 7 (5): 421–39. doi:10.3171/jns.1950.7.5.0421. PMID 14774761.