Peroxide
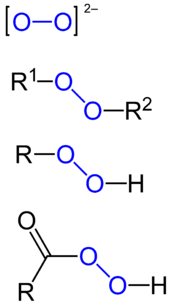
A peroxide is a compound containing an oxygen–oxygen single bond or the peroxide anion, O2−
2.[1] The O−O group is called the peroxide group or peroxo group. In contrast to oxide ions, the oxygen atoms in the peroxide ion have an oxidation state of −1.[2]
The simplest stable peroxide is hydrogen peroxide. Superoxides, dioxygenyls, ozones and ozonides are considered separately. Peroxide compounds can be roughly classified into organic and inorganic. Whereas the inorganic peroxides have an ionic, salt-like character, the organic peroxides are dominated by the covalent bonds. The oxygen–oxygen chemical bond of peroxide is unstable and easily split into reactive radicals via homolytic cleavage. For this reason, peroxides are found in nature only in small quantities, in water, atmosphere, plants, and animals. Peroxide ion formation has recently been highlighted as one of the main mechanisms by which oxides accommodate excess oxygen in ionic crystals and may have a large impact on a range of industrial applications including solid oxide fuel cells.[3][4]
Peroxides have a bleaching effect on organic substances and therefore are added to some detergents and hair colorants. Other large-scale applications include medicine and chemical industry, where peroxides are used in various synthesis reactions or occur as intermediate products. With an annual production of over 2 million tonnes, hydrogen peroxide is the most economically important peroxide. Many peroxides are unstable and hazardous substances; they cannot be stored and therefore are synthesized in situ and used immediately.
In biochemistry
Peroxides are usually very reactive and thus occur in nature only in a few forms. These include, in addition to hydrogen peroxide, a few vegetable products such as ascaridole and a peroxide derivative of prostaglandin. Hydrogen peroxide occurs in surface water, groundwater and in the atmosphere. It forms upon illumination or natural catalytic action by substances containing in water. Sea water contains 0.5 to 14 μg/L of hydrogen peroxide, freshwater 1 to 30 μg/L and air 0.1 to 1 parts per billion.[5]
Hydrogen peroxide is formed in human and animal organisms as a short-lived product in biochemical processes and is toxic to cells. The toxicity is due to oxidation of proteins, membrane lipids and DNA by the peroxide ions.[6] The class of biological enzymes called SOD (superoxide dismutase) is developed in nearly all living cells as an important antioxidant agent. They promote the disproportionation of superoxide into oxygen and hydrogen peroxide, which is then rapidly decomposed by the enzyme catalase to oxygen and water.[7]
- Formation of hydrogen peroxide by superoxide dismutase (SOD)
![{\displaystyle {\ce {{2O2^{-}}+2H+->[{\ce {SOD}}]\overbrace {H2O2} ^{hydrogen\ peroxide}+\overbrace {O2} ^{oxygen}}}}](../I/m/35d17b8365267e3819a1c1282879924cc5bfb9f1.svg)
Peroxisomes are organelles found in virtually all eukaryotic cells.[8] They are involved in the catabolism of very long chain fatty acids, branched chain fatty acids, D-amino acids, polyamines, and biosynthesis of plasmalogens, etherphospholipids critical for the normal function of mammalian brains and lungs.[9] Upon oxidation, they produce hydrogen peroxide in the following process:[10]
![{\displaystyle {\ce {{R-CH2-CH2-CO-SCoA}+O2->[{\ce {FAD}}]{R-CH=CH-CO-SCoA}+\overbrace {H2O2} ^{hydrogen\ peroxide}}}}](../I/m/9770f21e4fa0f6062860353b28f0d305fea95083.svg)
Catalase, another peroxisomal enzyme, uses this H2O2 to oxidize other substrates, including phenols, formic acid, formaldehyde, and alcohol, by means of the peroxidation reaction:
- , thus eliminating the poisonous hydrogen peroxide in the process.

This reaction is important in liver and kidney cells, where the peroxisomes neutralize various toxic substances that enter the blood. Some of the ethanol humans drink is oxidized to acetaldehyde in this way.[11] In addition, when excess H2O2 accumulates in the cell, catalase converts it to H2O through this reaction:
![{\displaystyle {\ce {\overbrace {H2O2} ^{hydrogen\ peroxide}->[{\ce {CAT}}]{1/2O2}+H2O}}}](../I/m/5b6ac46a8dad2afe5b4e98b4b0e9dc09a8fd2f2c.svg)
Another origin of hydrogen peroxide is the degradation of adenosine monophosphate which yields hypoxanthine. Hypoxanthine is then oxidatively catabolized first to xanthine and then to uric acid, and the reaction is catalyzed by the enzyme xanthine oxidase:[12]
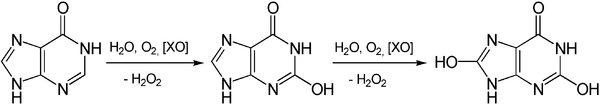


The degradation of guanosine monophosphate yields xanthine as an intermediate product which is then converted in the same way to uric acid with the formation of hydrogen peroxide.[12]
Eggs of sea urchin, shortly after fertilization by a sperm, produce hydrogen peroxide. It is then quickly dissociated to OH· radicals. The radicals serve as initiator of radical polymerization, which surrounds the eggs with a protective layer of polymer.[13]
The bombardier beetle has a device which allows it to shoot corrosive and foul-smelling bubbles at its enemies. The beetle produces and stores hydroquinone and hydrogen peroxide, in two separate reservoirs in the rear tip of its abdomen. When threatened, the beetle contracts muscles that force the two reactants through valved tubes into a mixing chamber containing water and a mixture of catalytic enzymes. When combined, the reactants undergo a violent exothermic chemical reaction, raising the temperature to near the boiling point of water. The boiling, foul-smelling liquid partially becomes a gas (flash evaporation) and is expelled through an outlet valve with a loud popping sound.[14][15][16]
Hydrogen peroxide is a signaling molecule of plant defense against pathogens.[17]
In firefly, oxidation of luciferins, which is catalyzed by luciferases, yields a peroxy compound 1,2-dioxetane. The dioxetane is unstable and decays spontaneously to carbon dioxide and excited ketones, which release excess energy by emitting light (bioluminescence).[18]

Hydrogen peroxide
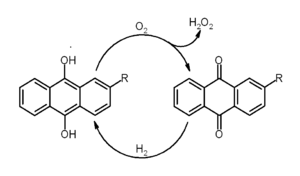
The most widely used synthesis method of hydrogen peroxide is the anthraquinone process. There, anthraquinone is first hydrogenated to anthrahydroquinone. This reduced compound is oxidized with molecular oxygen, regenerating anthraquinone and releasing hydrogen peroxide.[19] Direct synthesis of hydrogen peroxide from hydrogen and oxygen is inefficient and currently is not practiced industrially.[19]
Many peroxides of mineral acids, such as peroxodisulfates[20] and percarbonates,[21] can be obtained by anodic oxidation of the respective acids. The anode material must be stable to the required high potentials of a few volts and therefore is either platinum or its alloys.[22][23]

Peroxydisulfuric acid was historically used for the production of hydrogen peroxide in a method developed in the early 20th century:[5][19]

This process requires relatively high concentration of peroxydisulfuric acid as its more dilute solutions evolve oxygen gas instead of peroxide.[23]
Inorganic peroxides (aside from hydrogen peroxide)
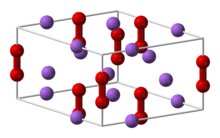
Inorganic peroxides are divided into ionic and covalent peroxide. The first class mostly contains the peroxides of the alkali and alkaline earth metals whereas the covalent peroxides are represented by such compounds as hydrogen peroxide and peroxymonosulfuric acid (H2SO5). In contrast to the purely ionic character of alkali metal peroxides, peroxides of transition metals have a more covalent character.[24] It should be noted that in some older literature some high-valence metal oxides are incorrectly named peroxides (e.g. PbO2, MnO2, Ag4O4) even though they do not contain the peroxide ion.
Defect formation in fluorite dioxides
Both CeO2 and ThO2 accommodate excess oxygen in their lattice by formaing peroxide molecules.[25] This is in contrast to UO2, which takes in excess oxygen by forming charged interstitials. The accommodation of excess oxygen in these systems changes their thermal and diffusion properties. This will have implications in the oxides' use in nuclear fuels and solid oxide fuel cells. Adding oxygen to the CeO2 system, for example, will increase the diffusivity of the oxygen allowing for more efficient solid oxide fuel cells.
Peroxide salts
Bonding in O2−
2

The peroxide ion is composed of two oxygen atoms that are linked by a single bond. The molecular orbital diagram of the peroxide dianion predicts a doubly occupied antibonding π* orbital and a bond order of one. The bond length is 149 pm, which is larger than in the ground state (triplet oxygen) of the oxygen molecule (3O2, 121 pm). This translates into the smaller force constant of the bond (2.8 N/cm vs. 11.4 N/cm for 3O2) and the lower frequency of the molecular vibration (770 cm−1 vs. 1555 cm−1 for 3O2).[26]
The peroxide ion can be compared with other molecular oxygen ions superoxide O−
2 and ozonide O−
3, but contrary to them, the peroxide is not a radical and not paramagnetic.[26] Owing to the weak bonding between the oxygen atoms, peroxide easily undergoes homolytic cleavage yielding two highly reactive radicals. This cleavage is accelerated by temperature, illumination or chemical reactions.

Preparation of peroxide salts
Most alkali metal peroxides can be synthesized directly by oxygenation of the elements. Lithium peroxide is formed upon treating lithium hydroxide with hydrogen peroxide:[24]
- 2 LiOH + H2O2 → Li2O2 + 2 H2O
Historically, barium peroxide is prepared by oxygenation of barium oxide at elevated temperature and pressure.[19]
- 2 BaO + O2 → 2 BaO2
Barium peroxide was once used to produce pure oxygen from air. This process relies on the temperature-dependent chemical balance between barium oxide and peroxide: the reaction of barium oxide with air at 500 °C results in barium peroxide, which upon heating to above 700 °C in oxygen decomposes back to barium oxide releasing pure oxygen.[19]
![{\displaystyle {\ce {\overbrace {2BaO} ^{barium\ oxide}+{O2}_{(air)}->[500^{\circ }{\ce {C}}]\overbrace {2BaO2} ^{barium\ peroxide}->[700^{\circ }{\ce {C}}]{2BaO}+{O2}_{(pure)}}}}](../I/m/88f5323ac6750a72be656a8a0e486244be8f2f08.svg)
Properties of peroxide salts
Few reactions are generally formulated for peroxide salt. In excess of dilute acids or water they release hydrogen peroxide.[24]
![{\displaystyle {\ce {{Na2O2}+2H2O->[{\ce {H3O^{+}}}]{2NaOH}+\overbrace {H2O2} ^{hydrogenperoxide}}}}](../I/m/d30801dc0cac1a5ae84340ccc222d391026da108.svg)
Upon heating, the reaction with water leads to the release of oxygen instead[24]
![{\displaystyle {\begin{array}{l}{\ce {{2Na2O2}+2H2O->{4NaOH}+O2}}\\{\ce {{2Na2O2}->[\Delta T]{2Na2O}+O2}}\end{array}}}](../I/m/3ae96f757f10776faf6070754330f4889e53a64b.svg)
The peroxide anion is a stronger nucleophile than hydroxide and displaces hydroxyl from oxyanions e.g. forming perborates and percarbonates. Sodium perborate and sodium percarbonate are important consumer and industrial bleaching agents; they stabilize hydrogen peroxide and limit side reactions (e.g. reduction and decomposition note below). The peroxide anion forms an adduct with urea, hydrogen peroxide–urea.
Hydrogen peroxide is both an oxidizing agent and reducing agent. The oxidation of hydrogen peroxide by sodium hypochlorite yields singlet oxygen. The net reaction of a ferric ion with hydrogen peroxide is a ferrous ion and oxygen. This proceeds via single electron oxidation and hydroxyl radicals. This is used in some organic chemistry oxidations, e.g. in the Fenton's reagent. Only catalytic quantities of iron ion is needed since peroxide also oxidizes ferrous to ferric ion. The net reaction of hydrogen peroxide and permanganate or manganese dioxide is manganous ion; however, until the peroxide is spent some manganous ions are reoxidized to make the reaction catalytic. This forms the basis for common monopropellant rockets.
As a ligand in coordination chemistry
Peroxide functions as a bidentate ligand in a variety of coordination complex. Some complexes have only peroxide ligands, e.g., chromium(VI) oxide peroxide (Cr(O
2)2−
4). Similarly, molybdate reacts in alkaline media with peroxide to form red peroxomolybdate Mo(O
2)2−
4.[27] The reaction of hydrogen peroxide with aqueous titanium(IV) gives a brightly colored peroxy complex that is a useful test for titanium as well as hydrogen peroxide. Many transition metal dioxygen complexes are best described as adducts of peroxide.[28]
Applications
Many inorganic peroxides are used for bleaching textiles and paper and as a bleaching additive to detergents and cleaning products.[19] The increasing environmental concerns resulted in the preference of peroxides over chlorine-based compounds and a sharp increase in the peroxide production.[5][29] The past use of perborates as additives to detergents and cleaning products[30] has been largely replaced by percarbonates in order to decrease the emission of boron to the environment. Sodium percarbonate is used in such products as OxiClean and Tide laundry detergent. When dissolved in water, it releases hydrogen peroxide and soda ash (sodium carbonate):[31]

The use of peroxide compounds in detergents is often reflected in their trade names; for example, Persil is a combination of the words perborate and silicate.
Some peroxide salts release oxygen upon reaction with carbon dioxide. This reaction is used in generation of oxygen from exhaled carbon dioxide on submarines and spaceships. Sodium or lithium peroxides are preferred in space applications because of their lower molar mass and therefore higher oxygen yield per unit weight.[19]

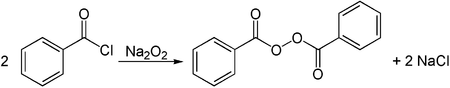
Alkali metal peroxides can be used for the synthesis of organic peroxides. One example is the conversion of benzoyl chloride with sodium peroxide to dibenzoyl peroxide.[32]
Organic peroxides
Organic peroxides can be divided into two major classes, peroxy acids and organic hydroperoxides. The first class is derived from the carboxylic acid and the second from ethers or alcohols.
Preparation
Most peroxy acids can be obtained by the reaction of hydrogen peroxide with the corresponding carboxylic acid:
- R = organic group

Another route employs acyl halides instead of the carboxylic acid. This method is used primarily with aromatic compounds under basic conditions in order to neutralize the resulting hydrogen chloride.

Aromatic aldehydes can be auto-oxidized into peroxycarboxylic acids:
- Ar = aryl

The products, however, react with the initial aldehyde forming the carboxylic acid:

Several synthesis routes are known for aliphatic peroxides, such as the reaction of dialkylsulfates with alkaline hydrogen peroxide solution.[33][34] In this method, the alkyl sulfate donates the alkyl group and the sulfate ion forms the leaving group.

This method can also yield cyclic peroxides.[35] The four-membered dioxetanes can be obtained by 2+2 cycloaddition of oxygen to alkenes.[36]
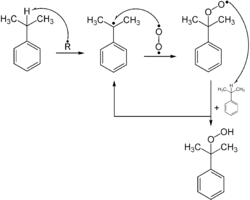
he selective synthesis of hydroperoxides can be carried out by free-radical oxidation of alkanes with oxygen. Here the active site formed by a radical initiator reacts with oxygen to form a hydroperoxyl. The addition of oxygen results in a more active radical which can further extract hydrogen atoms and release the hydroperoxide, leaving a new radical.[37] This process is used industrially for the synthesis of phenol from benzene and is called the Cumene process or Hock process for its cumene and cumene hydroperoxide intermediates.[38]
This auto-oxidation reaction can be used with common solvents from the group of ethers, such as diethyl ether, diisopropyl ether, tetrahydrofuran or 1,4-dioxane. It yields a volatile hydroperoxide ether that upon heating can result in a serious explosion.[37]

Peroxides are formed by living organisms through ene reactions or Diels–Alder reactions between alkenes and oxygen. Unsaturated fatty acids can serve as the olefinic substrates for the ene reaction and unsaturated amino acids like histidine can be the reactant for the Diels–Alder cyclization.[39] Rancidification (decomposition) of fats is partly caused by the formation of peroxides.
Properties and applications of organic peroxides
Peroxycarboxylic acids are generally weaker acids than the parent carboxylic acids. Like most peroxides, they are strong oxidants and tend to explode at high concentrations and higher temperatures.[37]
Organic peracids are used in the synthesis of epoxies via the Prilezhaev reaction.[40] Another important application is the synthesis of lactones of cyclic ketones in the Baeyer–Villiger oxidation process.[41] In both cases, electron-poor peroxycarboxylic acids are especially efficient, such as meta-chloroperoxybenzoic acid (mCPBA).


tert-Butyl hydroperoxide is a common oxidant in the Sharpless epoxidation, which is used for the stereoselective synthesis of epoxides. Karl Barry Sharpless was awarded the 2001 Nobel prize in Chemistry for this reaction.[42]
Peracetic acid is a popular disinfectant in the medical field and food industry.[43] Various peroxide solutions are commercially produced for the cleaning and disinfection of contact lenses.
Dibenzoyl peroxide is used as a radical initiator. Its weak peroxide bond cleaves easily yielding reactive benzoxy radicals, which assist polymerization leading to plastics like polyethylene.[44][45] One of the synthesis methods of the commercially important plastic caprolactam—the precursor to Nylon 6 (polycaprolactam)—is a Baeyer-Villiger rearrangement of cyclohexanone with peracetic acid. This yields caprolactone, which is then converted to caprolactam by reacting it with ammonia.[46]
Industrial resins based on acrylic and/or methacrylic acid esters are invariably produced by radical polymerization with organic peroxides at elevated temperatures.[47] The polymerization rate is adjusted by suitable choice of temperature and type of peroxide.[48]
Some peroxides are drugs, whose action is based on the formation of radicals at desired locations in the organism. For example, artemisinin and its derivatives, such as such artesunate, possess the most rapid action of all current drugs against falciparum malaria.[49] Artesunate is also efficient in reducing egg production in Schistosoma haematobium infection.[50]
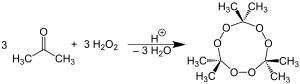
Many organic peroxides can initiate explosive polymerization in materials with unsaturated chemical bonds, and specifically triacetone triperoxide (TATP) and hexamethylene triperoxide diamine (HMTD) are powerful explosives. TATP is an inexpensive compound and is relatively easy to make. Whereas most other potent explosives, such as trinitrotoluene (TNT) or RDX (the major component of C4 mixtures), contain nitrogen, which is relatively easy to trace by sniffing techniques, TATP is nitrogen free and therefore is very difficult to detect by conventional screening methods. For this reason, it is an explosive favored by terrorists.[51] TATP and HMTD were used in several executed or planned terrorist acts of the early 2000s, most notably in the 2001 shoe bomb plot[52][53] and the 2005 London Underground bombings.[54][55] Several detection devices have been designed since those events. One, for example, releases a chemical mixture which changes color when interacting with traces of TATP.[53]
Laboratory identification

Several analytical methods are used for qualitative and quantitative determination of peroxides.[56] A simple qualitative detection of peroxides is carried out with the iodine-starch reaction.[57] Here peroxides, hydroperoxides or peracids oxidize the added potassium iodide into iodine, which reacts with starch producing a deep-blue color. Commercial paper indicators using this reaction are available. This method is also suitable for quantitative evaluation, but it can not distinguish between different types of peroxide compounds. Discoloration of various indigo dyes in presence of peroxides is used instead for this purpose.[58] For example, the loss of blue color in leuco-methylene blue is selective for hydrogen peroxide.[59]
Quantitative analysis of hydroperoxides is performed using potentiometric titration with lithium aluminium hydride.[60] Another way to evaluate the content of peracids and peroxides is the volumetric titration with alkoxides such as sodium ethoxide.[61]
Safety

Organic peroxides can accidentally initiate explosive polymerization in materials with unsaturated chemical bonds. Most notably, TATP and HMTD are high explosives, and TATP, because of its high susceptibility to accidental detonation by shock, friction, or sparks, has earned the nickname "Mother of Satan" among certain Islamic militant groups.[51] TATP can accidentally form as by-products in many commonly used reactions. These reactions range from synthesis of MDMA, where TATP is formed via isosafrole oxidation in acetone, to industrial production of phenol, where the second product of the cumene process, acetone, is partially oxidized to peroxide on the second reaction step. Accidental preparation of organic peroxides can occur by mixing ketone solvents (most commonly acetone) with waste materials containing hydrogen peroxide or other oxidizers and leaving the mixture standing for several hours. In addition, many liquid ethers in the presence of air, light and metals (which act as catalysts) slowly – over a period of months – form highly unstable ether peroxides such as diethyl ether peroxide. Therefore, ethers are often stored over potassium hydroxide, which not only destroys peroxides but also acts as a powerful desiccant.
Peroxides are also strong oxidizers and easily react with skin, cotton and wood pulp.[62] For safety reasons, peroxidic compounds are stored in a cool, opaque container, as heating and illumination accelerate their chemical reactions. Small amounts of peroxides, which emerge from storage or reaction vessels are neutralized using reducing agents such as iron(II) sulfate. The safety measures in industrial plants producing large amounts of peroxides include the following. The equipment is located within reinforced concrete structures with foil windows, which would relieve pressure and not shatter in case of explosion. The products are bottled in small containers and are moved to a cold place promptly after the synthesis. The containers are made of non-reactive materials such as stainless steel, some aluminium alloys or dark glass.[63]
History
Alexander von Humboldt synthesized one of the first synthetic peroxides, barium peroxide, in 1799 as a by-product of his attempts to decompose air. Nineteen years later Louis Jacques Thénard recognized that this compound could be used for the preparation of a previously unknown compound, which he described as oxidized water – subsequently known as hydrogen peroxide.[64] Thénard and Joseph Louis Gay-Lussac synthesized sodium peroxide in 1811. The bleaching effect of peroxides and their salts on natural dyes became known around that time, but early attempts of industrial production of peroxides failed, and the first plant producing hydrogen peroxide was built in 1873 in Berlin. The discovery of the synthesis of hydrogen peroxide by electrolysis with sulfuric acid introduced the more efficient electrochemical method. It was first implemented into industry in 1908 in Weißenstein, Carinthia, Austria. The anthraquinone process, which is still used, was developed during the 1930s by the German chemical manufacturer IG Farben in Ludwigshafen. The increased demand and improvements in the synthesis methods resulted in the rise of the annual production of hydrogen peroxide from 35,000 tonnes in 1950, to over 100,000 tonnes in 1960, to 300,000 tonnes by 1970; by 1998 it reached 2.7 million tonnes.[5]
See also
References
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "peroxides".
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "oxidation state".
- ↑ Middleburgh S. C., Lagerlof K.P.D. and Grimes R.W. (2013). "Accommodation of Excess Oxygen in Group II Oxides". Journal of the American Ceramic Society. 96: 308. doi:10.1111/j.1551-2916.2012.05452.x.
- ↑ S. C. Middleburgh, I. Karatchevtseva, B. J. Kennedy, P. A. Burr, Z. Zhang, E. Reynolds, R. W. Grimes and G. R. Lumpkin (2014). "Peroxide defect formation in zirconate perovskites". Journal of Materials Chemistry A. doi:10.1039/c4ta02558j.
- 1 2 3 4 Offermanns, Heribert; Dittrich, Gunther; Steiner, Norbert (2000). "Wasserstoffperoxid in Umweltschutz und Synthese". Chemie in unserer Zeit. 34 (3): 150. doi:10.1002/1521-3781(200006)34:3<150::AID-CIUZ150>3.0.CO;2-A.
- ↑ Löffler G. and Petrides, P. E. Physiologische Chemie. 4 ed., p. 288, Springer, Berlin 1988, ISBN 3-540-18163-6 (in German)
- ↑ Löffler G. and Petrides, P. E. Physiologische Chemie. 4 ed., pp. 321–322, Springer, Berlin 1988, ISBN 3-540-18163-6 (in German)
- ↑ Gabaldón T (2010). "Peroxisome diversity and evolution". Philos Trans R Soc Lond B Biol Sci. 365 (1541): 765–73. doi:10.1098/rstb.2009.0240. PMC 2817229
 . PMID 20124343.
. PMID 20124343. - ↑ Wanders RJ, Waterham HR (2006). "Biochemistry of mammalian peroxisomes revisited". Annu. Rev. Biochem. 75 (1): 295–332. doi:10.1146/annurev.biochem.74.082803.133329. PMID 16756494.
- ↑ Nelson, David; Cox, Michael; Lehninger, Albert L. and Cox, Michael M. Lehninger Biochemie, pp. 663–664, Springer, 2001, ISBN 3-540-41813-X (in German)
- ↑ Riley, Edward P. et al. (ed.) Fetal Alcoholspectrum Disorder Fasd: Management and Policy Perspectives, Wiley-VCH, 2010, ISBN 3-527-32839-4 p. 112
- 1 2 Nelson, David; Cox, Michael; Lehninger, Albert L. and Cox, Michael M. Lehninger Biochemie, p. 932, Springer, 2001, ISBN 3-540-41813-X (in German)
- ↑ Kröger, M. (1989). "History". Chemie in unserer Zeit. 23: 34. doi:10.1002/ciuz.19890230106.
- ↑ Schildknecht, H.; Holoubek, K. (1961). "The bombardier beetle and its chemical explosion". Angewandte Chemie. 73: 1. doi:10.1002/ange.19610730102.
- ↑ Weber CG (Winter 1981). "The Bombadier Beetle Myth Exploded". Creation/Evolution. National Center for Science Education. 2 (1): 1–5.
- ↑ Isaak, Mark (May 30, 2003). "Bombardier Beetles and the Argument of Design". TalkOrigins Archive.
- ↑ Wie Pflanzen sich schützen, Helmholtz-Institute of Biochemical Plant Pathology (in German)
- ↑ Aldo Roda Chemiluminescence and Bioluminescence: Past, Present and Future, p. 57, Royal Society of Chemistry, 2010, ISBN 1-84755-812-7
- 1 2 3 4 5 6 7 Wiberg, Egon; Wiberg, Nils and Holleman, Arnold Frederick Inorganic Chemistry, Academic Press, 2001, ISBN 0-12-352651-5, pp. 471–502
- ↑ Jander, Blasius, Strähle Einführung in das anorganisch-chemische Praktikum. 14ed., pp. 311–312, Hirzel Verlag, Stuttgart, 1995, ISBN 978-3-7776-0672-9
- ↑ Riesenfeld, E. H.; Reinhold, B. (1909). "The existence of real percarbonates and their distinction from carbonates with hydrogen peroxide-crystal". Berichte der deutschen chemischen Gesellschaft. 42 (4): 4377. doi:10.1002/cber.19090420428.
- ↑ Hamann, Carl H.; Hamnett, A. and Vielstich, Wolf Electrochemistry, Wiley-VCH, 1998, ISBN 3-527-29095-8, p. 320
- 1 2 Serrano, K; Michaud, P.A.; Comninellis, C.; Savall, A. (2002). "Electrochemical preparation of peroxodisulfuric acid using boron doped diamond thin film electrodes" (PDF). Electrochimica Acta. 48 (4): 431. doi:10.1016/S0013-4686(02)00688-6.
- 1 2 3 4 Vol'nov, I. I. Peroxides, superoxides and ozonides of alkali and alkaline earth metals, pp. 21–51, Plenum Press, New York, 1966, no ISBN
- ↑ Middleburgh S. C., Lumpkin G.R. and Grimes R.W. (2013). "Accommodation of Excess Oxygen in Fluorite Dioxides". Solid State Ionics. 253: 119–122. doi:10.1016/j.ssi.2013.09.020.
- 1 2 Wiberg, Egon; Wiberg, Nils and Holleman, Arnold Frederick Inorganic Chemistry, Academic Press, 2001, ISBN 0-12-352651-5, pp. 475 ff
- ↑ Eagleson, Mary (1994). Concise encyclopedia chemistry. Walter de Gruyter. pp. 660–. ISBN 978-3-11-011451-5.
- ↑ Greenwood, N. N.; & Earnshaw, A. (1997). Chemistry of the Elements (2nd Edn.), Oxford:Butterworth-Heinemann. ISBN 0-7506-3365-4.
- ↑ Ullmann's Encyclopedia of Industrial Chemistry, Vol A 19, 5 ed., pp. 177–197, VCH, Weinheim, 1991, ISBN 3-527-20138-6
- ↑ Brotherton, B.J. "Boron: Inorganic Chemistry", in Encyclopedia of Inorganic Chemistry (1994) Ed. R. Bruce King, John Wiley & Sons ISBN 0-471-93620-0
- ↑ Jones, Craig W. (1999). Applications of hydrogen peroxide and its derivatives. Royal Society of Chemistry. ISBN 0-85404-536-8.
- ↑ Gambarjan, Stephan (1909). "Diphenylamine and Acylperoxyde". Berichte der deutschen chemischen Gesellschaft. 42 (3): 4003. doi:10.1002/cber.190904203164.
- ↑ Medwedew, S. S.; Alexejewa, E. N. (1932). "Organic peroxides II. Of the reaction between benzoyl hydroperoxide or benzoyl peroxide and triphenylmethyl". Berichte der deutschen chemischen Gesellschaft (A and B Series). 65 (2): 137. doi:10.1002/cber.19320650204.
- ↑ Wiley, Richard Haven "Preparation of diaikyl peroxides" U.S. Patent 2,357,298 Issue date: 1942
- ↑ Criegee, Rudolf; Müller, Gerhard (1956). "1.2-Dioxan". Chemische Berichte. 89 (2): 238. doi:10.1002/cber.19560890209.
- ↑ Heinz G. O. Becker Organikum, Wiley-VCH, 2001, ISBN 3-527-29985-8, p. 323
- 1 2 3 Heinz G. O. Becker Organikum, Wiley-VCH, 2001, ISBN 3-527-29985-8 pp. 206–207
- ↑ Brückner, R. Reaktionsmechanismen: organische Reaktionen, Stereochemie, moderne Synthesemethoden, pp. 41–42, Spektrum Akademischer Verlag, Munich, 2004, ISBN 3-8274-1579-9 (in German)
- ↑ Autor, Anne Pomeroy Pathology of Oxygen, pp. 25–26, Academic Press, New York, 1982, ISBN 0-12-068620-1
- ↑ Beyer, Hans; Walter, Wolfgang and Francke, Wittko Lehrbuch der organischen Chemie, 23 Ed., pp. 76–77, Hirzel Verlag, Stuttgart-Leipzig, 1998, ISBN 3-7776-0808-4
- ↑ Vollhardt, Kurt Peter C. and Schore, Neil Eric Organische Chemie, 3rd ed., pp. 818–819, Wiley-VCH, 2000, ISBN 3-527-29819-3
- ↑ Bülle, Jan and Hüttermann, Aloys Das Basiswissen der organischen Chemie, pp. 308–309, Wiley-VCH Weinheim, 2000, ISBN 3-527-30847-4
- ↑ Block, S. S. Disinfection, Sterilization and Preservation, Lippincott, Williams & Wilkins, Philadelphia, 2000, ISBN 0-683-30740-1, p. 979
- ↑ Frankenburg, W. G. Advances in Catalysis and Related Subjects, Vol. 2, pp. 24–26, 1948, Academic Press, ISBN 0-12-007801-5
- ↑ Wicks, Z. W.; Jones, F. N.; Pappas, S. P. and Wicks, D. A. Organic Coatings, 3rd edition, pp. 17–26, 2007, Wiley, New York, ISBN 0-471-69806-7
- ↑ Arpe, H.–J. Industrielle Organische Chemie: Bedeutende Vor- und Zwischenprodukte, pp. 284–285, Wiley-VCH Weinheim, 2007, ISBN 3-527-31540-3
- ↑ Thomas Brock, Michael Groteklaes, Peter Mischke Lehrbuch der Lacktechnologie, Vincentz Network GmbH & Co KG, 2000, ISBN 3-87870-569-7 p. 67
- ↑ Organische Peroxide für die Polymerisation. pergan.com (in German)
- ↑ White NJ (1997). "Assessment of the pharmacodynamic properties of antimalarial drugs in vivo". Antimicrob. Agents Chemother. 41 (7): 1413–22. PMC 163932. PMID 9210658.
- ↑ Boulangier D, Dieng Y, Cisse B, et al. (2007). "Antischistosomal efficacy of artesunate combination therapies administered as curative treatments for malaria attacks". Trans R Soc Trop Med Hyg. 101 (2): 113–16. doi:10.1016/j.trstmh.2006.03.003. PMID 16765398.
- 1 2 Genuth, Iddo & Fresco-Cohen, Lucille (6 November 2006). "TATP: Countering the Mother of Satan". The Future of Things. Retrieved 24 September 2009.
The tremendous devastative force of TATP, together with the relative ease of making it, as well as the difficulty in detecting it, made TATP one of the weapons of choice for terrorists
- ↑ "Judge denies bail to accused shoe bomber". CNN. December 28, 2001.
- 1 2 "Terrorist Use Of TATP Explosive". Opensourcesinfo.org. 2005-07-25. Retrieved 2010-01-18.
- ↑ "The real story of 7/7", The Observer, May 7, 2006
- ↑ London bombers used everyday materials—U.S. police, Reuters, 4 August 2005
- ↑ Légrádi, L.; Légrádi, J. (1970). "Detection of peroxides, hydroperoxides and peracids". Mikrochimica Acta. 58: 119. doi:10.1007/BF01218105.
- ↑ Lea, C. H. (1931). "The Effect of Light on the Oxidation of Fats". Proceedings of the Royal Society B: Biological Sciences. 108 (756): 175. doi:10.1098/rspb.1931.0030.
- ↑ Veibel, S. Analytik organischer Verbindungen, Akademie-Verlag, Berlin, 1960, p. 262
- ↑ Eiss, M. I.; Giesecke, Paul (1959). "Colorimetric Determination of Organic Peroxides". Analytical Chemistry. 31 (9): 1558. doi:10.1021/ac60153a038.
- ↑ Higuchi, T.; Zuck, Donald Anton (1951). "Behaviors of Several Compounds as Indicators in Lithium Aluminum Hydride Titration of Functional Groups". Journal of the American Chemical Society. 73 (6): 2676. doi:10.1021/ja01150a073.
- ↑ Martin, A. J. (1957). "Potentiometric titration of hydroperoxide and peracid in Anhydrous Ethylenediamine". Analytical Chemistry. 29: 79. doi:10.1021/ac60121a022.
- ↑ Heinz G. O. Becker Organikum, Wiley-VCH, 2001, ISBN 3-527-29985-8 pp. 741–762
- ↑ Ozonelab Peroxide compatibility
- ↑ Gilbert, L. W. (1820). "Der tropfbar flüssige Sauerstoff, oder das oxygenierte Wasser". Annals of Physics (in German). 65–66: 3. doi:10.1002/andp.18200640102.