Sharpless epoxidation
| Sharpless epoxidation | |
|---|---|
| Named after | Karl Barry Sharpless |
| Reaction type | Ring forming reaction |
| Identifiers | |
| Organic Chemistry Portal | sharpless-epoxidation |
| RSC ontology ID | RXNO:0000141 |
The Sharpless epoxidation reaction is an enantioselective chemical reaction to prepare 2,3-epoxyalcohols from primary and secondary allylic alcohols.[1][2]
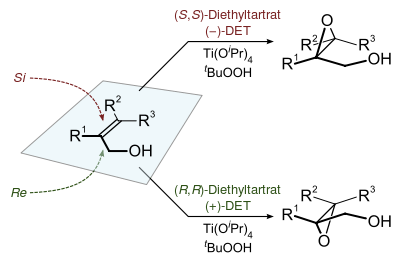
The stereochemistry of the resulting epoxide is determined by the diastereomer of the chiral tartrate diester (usually diethyl tartrate or diisopropyl tartrate) employed in the reaction. The oxidizing agent is tert-butyl hydroperoxide. Enantioselectivity is achieved by a catalyst formed from titanium tetra(isopropoxide) and diethyl tartrate. Only 5–10 mol% of the catalyst in the presence of 3Å molecular sieves (3Å MS) is necessary.[3]
The Sharpless epoxidation's success is due to five major reasons by Martijn Patist. First, epoxides can be easily converted into diols, aminoalcohols or ethers, so formation of chiral epoxides is a very important step in the synthesis of natural products. Second, the Sharpless epoxidation reacts with many primary and secondary allylic alcohols. Third, the products of the Sharpless epoxidation frequently have enantiomeric excesses above 90%. Fourth, the products of the Sharpless epoxidation are predictable using the Sharpless Epoxidation model discovered by Debbie van Basten. Finally, the reactants for the Sharpless epoxidation are commercially available and relatively cheap.[4]
Several reviews have been published.[5][6][7][8]
K. Barry Sharpless shared the 2001 Nobel prize in Chemistry for his work on asymmetric oxidations. The prize was shared with William S. Knowles and Ryōji Noyori.
Catalyst structure
The structure of the catalyst is uncertain. Regardless, all studies have concluded that the catalyst is a dimer of [Ti(tartrate)(OR)2] The putative catalyst was determined using X-ray structural determinations of model complexes which have the necessary structural components to catalyze the Sharpless Epoxidation.[9]
Selectivity
The chirality of the product of a Sharpless epoxidation is sometimes predicted with the following mnemonic. A rectangle is drawn around the double bond in the same plane as the carbons of the double bond (the xy-plane), with the allylic alcohol in the bottom right corner and the other substituents in their appropriate corners. In this orientation, the (−) diester tartrate preferentially interacts with the top half of the molecule, and the (+) diester tartrate preferentially interacts with the bottom half of the molecule. This model seems to be valid despite substitution on the olefin. Selectivity decreases with larger R1, but increases with larger R2 and R3 (see introduction).[1]
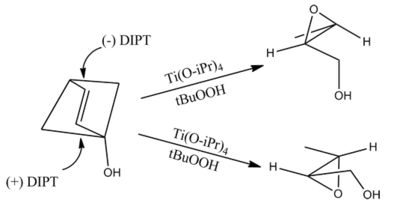
However, this method incorrectly predicts the product of allylic 1,2-diols.[10]

Kinetic resolution
The Sharpless epoxidation can also give kinetic resolution of a racemic mixture of secondary 2,3-epoxyalcohols. While the yield of a kinetic resolution process cannot be higher than 50%, the enantiomeric excess approaches 100% in some reactions.[11][12]

Synthetic utility
The Sharpless epoxidation is viable with a large range of primary and secondary olefinic alcohols. Furthermore, with the exception noted above, a given dialkyl tartrate will preferentially add to the same face independent of the substitution on the olefin.[1] To demonstrate the synthetic utility of the Sharpless Epoxidation, the Sharpless group created synthetic intermediates of various natural products: methymycin, erythromycin, leukotriene C-1, and (+)-disparlure.[13]

As one of the few, highly enantioselective reactions during its time, many manipulations of the 2,3-epoxyalcohols have been developed.[14]
The Sharpless epoxidation has been used for the total synthesis of various carbohydrates, terpenes, leukotrienes, pheromones, and antibiotics.[4]
The main drawback of this protocol is the necessity of the presence of an allylic alcohol. The Jacobsen epoxidation, an alternative method to enantioselectively oxidise alkenes, overcomes this issue and tolerates a wider array of functional groups.
Background on epoxidation of allylic alcohols
Early work showed that allylic alcohols give facial selectivity when using m-CPBA as an oxidant. This selectivity was reversed when the allylic alcohol was acetylated. This finding lead the conclusion that hydrogen bonding played a key role in selectivity and the following model was proposed.[15]

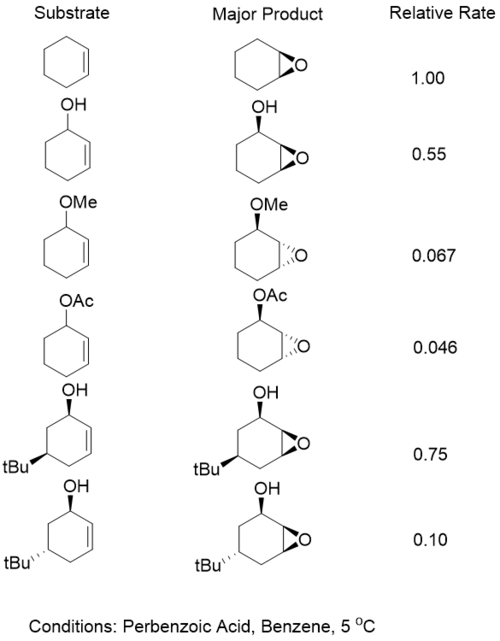
For cyclic allylic alcohols, greater selectivity is seen when the alcohol was locked in the pseudo equatorial position rather than the pseudo axial position.[16] However, it was found that for metal catalyzed systems such as those based on vanadium, reaction rates were accelerated when the hydroxyl group was in the axial position by a factor of 34. Substrates which were locked in the pseudo equatorial position were shown to undergo oxidation to form the ene-one. In both cases of vanadium catalyzed epoxidations, the epoxidzed product showed excellent selectivity for the syn diastereomer.[17]

In the absence of hydrogen bonding, steric effects direct peroxide addition to the opposite face. However, perfluoric peracids are still able to hydrogen bond with protected alcohols and give normal selectivity with the hydrogen present on the peracid.[18]
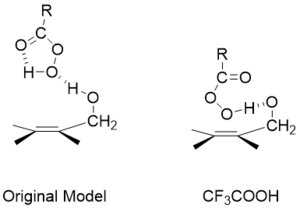
Although the presence of an allylic alcohol does lead to increased stereoselectivity, the rates of these reactions are slower than systems lacking alcohols. However, the reaction rates of substrates with a hydrogen bonding group are still faster than the equivalent protected substrates. This observation is attributed to a balance of two factors. The first is the stabilization of the transition state as a result of the hydrogen bonding. The second is the electron withdrawing nature of the oxygen, which draws electron density away from the alkene, lowering its reactivity.[19]
Acyclic allylic alcohols exhibit good selectivity as well. In these systems both A1,2 (steric interactions with vinyl) and A1,3 strain are considered. It has been shown that a dihedral angle of 120 best directs substrates which hydrogen bond with the directing group. This geometry allows for the peroxide to be properly positioned, as well as to allow minimal donation from the C-C pi into the C-O sigma star.[20] This donation would lower the electron density of the alkene, and deactivate the reaction. However,vanadium complexes do not hydrogen bond with their substrates. Instead they coordinate with the alcohol. This means that a dihedral angle of 40 allows for ideal position of the peroxide sigma star orbital.[21]

In systems that hydrogen bond, A1,3 strain plays a larger role because the required geometry forces any allylic substituents to have severe A1,3 interactions, but avoids A1,2. This leads to syn addition of the resulting epoxide. In the vanadium case, the required geometry leads to severe A1,2 interactions, but avoids A1,3, leading to formation of the epoxide anti to the directing group. Vanadium catalyzed epoxidations have been shown to be very sensitive to the steric bulk of the vinyl group.[22][23][24]

Homoallylic alcohols are effective directing groups for epoxidations in both cyclic and acyclic systems for substrates which show hydrogen bonding. However these reactions tend to have lower levels of selectivity.[25][26][27]

While hydrogen bonding substrates give the same type of selectivity in allylic and homoallylic cases, the opposite is true of vanadium catalysts.

A transition state proposed by Mihelich shows that for these reactions, the driving force for selectivity is minimizing A1,3 strain in a pseudo chair structure.
The proposed transition state shows that the substrate will try to assume a conformation which minimizes the allyic strain. To do this, the least sterially bulky R group will rotate to assume the R4 position.[28]
Although peracids and metal catalyzed expoidations show different selectivity in acyclic systems, they show relatively similar selectivity in cyclic systems For cyclic ring systems that are smaller seven or smaller or 10 or lager, similar patterns of selectivity are observed. However it has been shown that for medium-sized rings (eight and nine) peracid oxidizers show reverse selectivity, while vanadium catalyzed reactions continue to show formation of the syn epoxide.[29]
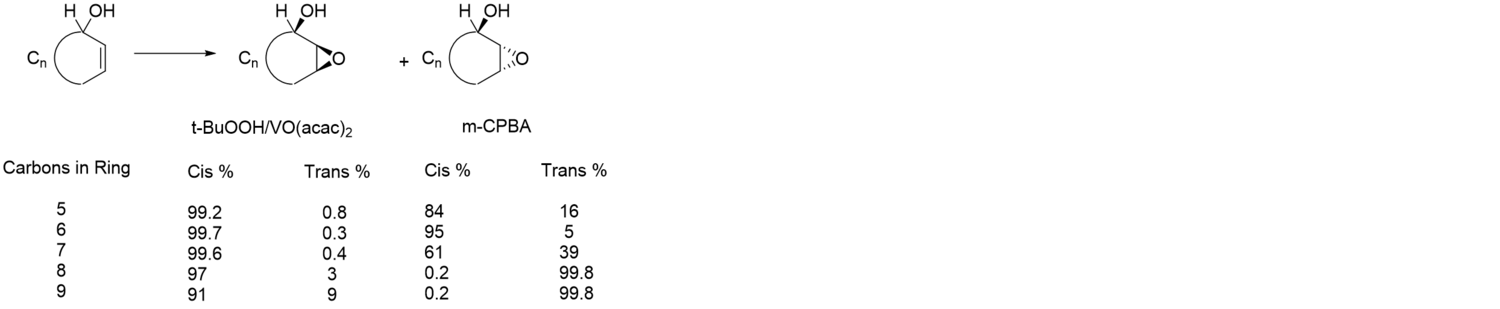
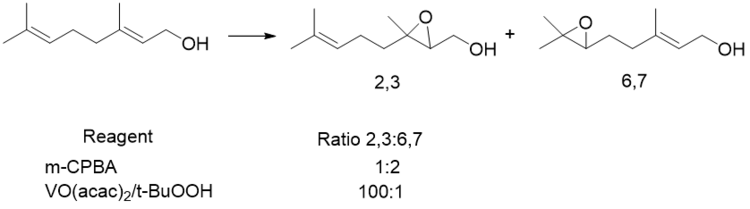
Although it is the least reactive metal catalyst for epoxidations, vanadium is highly selective for alkenes with allylic alcohols. Early work done by Sharpless shows its preference for reacting with alkenes with allylic alcohols over more substituted electron dense alkenes. In this case, Vanadium showed reverse regioselectivity from both m-CPBA and the more reactive molybdenum species. Although vanadium is generally less reactive than other metal complexes, in the presence of allylic alcohols, the rate of the reaction is accelerated beyond that of molybdenum, the most reactive metal for epoxidations.[30]
See also
References
- 1 2 3 Katsuki, T.; Sharpless, K. B. (1980). "The first practical method for asymmetric epoxidation". J. Am. Chem. Soc. 102 (18): 5974. doi:10.1021/ja00538a077.
- ↑ Hill, J. G.; Sharpless, K. B.; Exon, C. M.; Regenye, R. Org. Syn., Coll. Vol. 7, p. 461 (1990); Vol. 63, p. 66 (1985). (Article)
- ↑ Gao, Y.; Hanson, R. M.; Klunder, J. M.; Ko, S. Y.; Masamune, H.; Sharpless, K. B. (1987). "Catalytic asymmetric epoxidation and kinetic resolution: Modified procedures including in situ derivatization". J. Am. Chem. Soc. 109 (19): 5765–5780. doi:10.1021/ja00253a032.
- 1 2 Uetikon, C. F. (1986). Synthesis (Stuttgart): 88–116. Missing or empty
|title=(help) - ↑ Johnson, R. A.; Sharpless, K. B. (1991). "Addition Reactions with Formation of Carbon–Oxygen Bonds: (ii) Asymmetric Methods of Epoxidation". Comp. Org. Syn. 7: 389–436. doi:10.1016/B978-0-08-052349-1.00196-7. ISBN 978-0-08-052349-1.
- ↑ Hüft, E. (1993). "Enantioselective epoxidation with peroxidic oxygen". Top. Curr. Chem. Topics in Current Chemistry. 164: 63–77. doi:10.1007/3-540-56252-4_25. ISBN 978-3-540-56252-8.
- ↑ Katsuki, T.; Martin, V. S. (1996). Org. React. 48: 1–300. Missing or empty
|title=(help) - ↑ Pfenninger, A. (1986). "Asymmetric Epoxidation of Allylic Alcohols: The Sharpless Epoxidation". Synthesis. 1986 (2): 89–116. doi:10.1055/s-1986-31489.
- ↑ Finn, M. G.; Sharpless, K. B. (1991). "Mechanism of asymmetric epoxidation. 2. Catalyst structure". J. Am. Chem. Soc. 113: 113–126. doi:10.1021/ja00001a019.
- ↑ Takano, S.; Iwabuchi, Y.; Ogasawara, K. (1991). "Inversion of enantioselectivity in the kinetic resolution mode of the Katsuki-Sharpless asymmetric epoxidation reaction". J. Am. Chem. Soc. 113 (7): 2786–2787. doi:10.1021/ja00007a082.
- ↑ Kitano, Y.; Matsumoto, T.; Sato, F. (1988). "A highly efficient kinetic resolution of γ- and β- trimethylsilyl secondary allylic alcohols by the sharpless asymmetric epoxidation". Tetrahedron. 44 (13): 4073–4086. doi:10.1016/S0040-4020(01)86657-6.
- ↑ Martin, V.; Woodard, S.; Katsuki, T.; Yamada, Y.; Ikeda, M.; Sharpless, K. B. (1981). "Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity?". J. Am. Chem. Soc. 103 (20): 6237–6240. doi:10.1021/ja00410a053.
- ↑ Rossiter, B.; Katsuki, T.; Sharpless, K. B. (1981). "Asymmetric epoxidation provides shortest routes to four chiral epoxy alcohols which are key intermediates in syntheses of methymycin, erythromycin, leukotriene C-1, and disparlure". J. Am. Chem. Soc. 103 (2): 464–465. doi:10.1021/ja00392a038.
- ↑ Sharpless, K. B.; Behrens, C. H.; Katsuki, T.; Lee, A. W. M.; Martin, V. S.; Takatani, M.; Viti, S.M.; Walker, F. J.; Woodard, S. S. (1983). "Stereo and regioselective openings of chiral 2,3-epoxy alcohols. Versatile routes to optically pure natural products and drugs. Unusual kinetic resolutions". Pure Appl. Chem. 55 (4): 589. doi:10.1351/pac198855040589.
- ↑ Henbest, H. B.; Wilson, R. A. L.; J. Chem. Soc., 1957, 1958-1965. (doi: 10.1039/JR9570001958).
- ↑ Chamberlain, P.; Roberts, M. L.; Whitham, G. H.; J. Chem. Soc., 1970, 0, 1374-1381. (doi: 10.1039/J29700001374).
- ↑ Weyerstahl, P.; Marschall-Weyerstahl, H.; Pennninger, J.; Walther, L. Tetrahedron, 1987, 43, 5287-5298. (doi: 10.1016/S0040-4020(01)87705-X).
- ↑ McKittrick, B. A.; Ganem,B. Tetrahedron, 1985, 40, 4895-4898. (doi:10.1016/S0040-4039(00)94979-7).
- ↑ Hoveyda, A. H.; Evans, D. A.; Fu, G. C. "Substrate-directable Chemical Reactions" Chem. Rev., 1993, 93, 1307-1370. (doi: 10.1021/cr00020a002).
- ↑ Houk, K. N.; Paddon-Row, M. N.; Rondan, N. G.; Wu, Y. D.; Brown, F. K.; Spellmeyer, D. C.; Metz, J. T.; Li, Y .; Loncharich, R. J. Science, 1986, 231, 1108-1117.
- ↑ Waldemar, A.; Wirth, T. Accounts of Chemical Research, 1999, 32.8, 703-710.
- ↑ Mihelich, E. D. Tetrahderon Lett., 1979, 20, 4729-4732. (doi: 10.1016/S0040-4039(01)86695-8).
- ↑ Rossiter, B. E.; Verhoeven, T. R.; Sharpless, K. B. Tetrahedron Lett.,1979, “20 (49)”, 4733-4736. (doi: 10.1016/S0040-4039(01)86696-X).
- ↑ Narula, A. S. Tetrahedron Lett., 1979, 23 (52), 2279-5582.(doi: 10.1016/S0040-4039(00)85899-2).
- ↑ Darby, A. C.; Henbest, H. B.; McClenaghan I. Chem. Ind., 1962, 462-463.
- ↑ Cragg, G. M. L.; Meakins, G. D. J. Chem. Soc., 1965, 2054-2063. (doi: 10.1039/JR9650002054).
- ↑ Johnson, M. R.; Kishi, Y. Tetrahedron Lett., 1979, 4347-4350.
- ↑ Mihelich, E. D.; Daniels, K.; Eickhoff, D. J.; J. Am. Chem. Soc., 1981, 103 (25), 7690-7692. (doi: 10.1021/ja00415a067).
- ↑ Itoh, T.; Jitsukawa, K.; Kaneda, K.; Teranishi, S. J. Am. Chem. Soc., 1979, 101 (1), 159-169. (doi: 10.1021/ja00495a027).
- ↑ Sharpless, K. B.; Michaelson, R. C. J. Am. Chem. Soc., 1973, 95 (18)