Base excision repair

In biochemistry and genetics, base excision repair (BER) is a cellular mechanism that repairs damaged DNA throughout the cell cycle. It is responsible primarily for removing small, non-helix-distorting base lesions from the genome. The related nucleotide excision repair pathway repairs bulky helix-distorting lesions. BER is important for removing damaged bases that could otherwise cause mutations by mispairing or lead to breaks in DNA during replication. BER is initiated by DNA glycosylases, which recognize and remove specific damaged or inappropriate bases, forming AP sites. These are then cleaved by an AP endonuclease. The resulting single-strand break can then be processed by either short-patch (where a single nucleotide is replaced) or long-patch BER (where 2-10 new nucleotides are synthesized).[1]
Lesions processed by BER
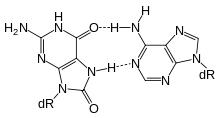
Single bases in DNA can be chemically damaged by a variety of mechanisms, the most common ones being deamination, oxidation, and alkylation. These modifications can affect the ability of the base to hydrogen-bond, resulting in incorrect base-pairing, and, as a consequence, mutations in the DNA. For example, incorporation of adenine across from 8-oxoguanine (right) during DNA replication causes a G:C base pair to be mutated to T:A. Other examples of base lesions repaired by BER include:
- Oxidized bases: 8-oxoguanine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG, FapyA)
- Alkylated bases: 3-methyladenine, 7-methylguanosine
- Deaminated bases: hypoxanthine formed from deamination of adenine. Xanthine formed from deamination of guanine. (Thymidine products following deamination of 5-methylcytosine are more difficult to recognize, but can be repaired by mismatch-specific glycosylases)
- Uracil inappropriately incorporated in DNA or formed by deamination of cytosine[2]
In addition to base lesions, the downstream steps of BER are also utilized to repair single-strand breaks.
The choice between long-patch and short-patch repair
The choice between short- and long-patch repair is currently under investigation. Various factors are thought to influence this decision, including the type of lesion, the cell cycle stage, and whether the cell is terminally differentiated or actively dividing.[3] Some lesions, such as oxidized or reduced AP sites, are resistant to pol β lyase activity and, therefore, must be processed by long-patch BER.
Pathway preference may differ between organisms, as well. While human cells utilize both short- and long-patch BER, the yeast Saccharomyces cerevisiae was long thought to lack a short-patch pathway because it does not have homologs of several mammalian short-patch proteins, including pol β, DNA ligase III, XRCC1, and the kinase domain of PNKP. The recent discovery that the poly-A polymerase Trf4 possesses 5' dRP lyase activity has challenged this view.[4]
Proteins involved in base excision repair
DNA glycosylases
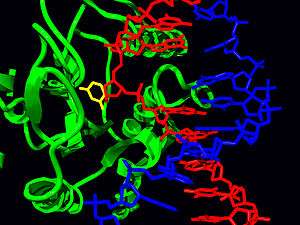
DNA glycosylases are responsible for initial recognition of the lesion. They flip the damaged base out of the double helix, as pictured, and cleave the N-glycosidic bond of the damaged base, leaving an AP site. There are two categories of glycosylases: monofunctional and bifunctional. Monofunctional glycosylases have only glycosylase activity, whereas bifunctional glycosylases also possess AP lyase activity. Therefore, bifunctional glycosylases can convert a base lesion into a single-strand break without the need for an AP endonuclease. β-Elimination of an AP site by a glycosylase-lyase yields a 3' α,β-unsaturated aldehyde adjacent to a 5' phosphate, which differs from the AP endonuclease cleavage product.[5] Some glycosylase-lyases can further perform δ-elimination, which converts the 3' aldehyde to a 3' phosphate. A wide variety of glycosylases have evolved to recognize different damaged bases. Examples of DNA glycosylases include Ogg1, which recognizes 8-oxoguanine, Mag1, which recognizes 3-methyladenine, and UNG, which removes uracil from DNA.
AP endonucleases
The AP endonucleases cleave an AP site to yield a 3' hydroxyl adjacent to a 5' deoxyribosephosphate (dRP). AP endonucleases are divided into two families based on their homology to the ancestral bacterial AP endonucleaes endonuclease IV and exonuclease III.[6] Many eukaryotes have members of both families, including the yeast Saccharomyces cerevisiae, in which Apn1 is the EndoIV homolog and Apn2 is related to ExoIII. In humans, two AP endonucleases, APEX1 and APEX2, have been identified.[7] It is a member of the ExoIII family.
End processing enzymes
In order for ligation to occur, a DNA strand break must have a hydroxyl on its 3' end and a phosphate on its 5' end. In humans, polynucleotide kinase-phosphatase (PNKP) promotes formation of these ends during BER. This protein has a kinase domain, which phosphorylates 5' hydroxyl ends, and a phosphatase domain, which removes phosphates from 3' ends. Together, these activities ready single-strand breaks with damaged termini for ligation. The AP endonucleases also participate in 3' end processing. Besides opening AP sites, they possess 3' phosphodiesterase activity and can remove a variety of 3' lesions including phosphates, phosphoglycolates, and aldehydes. 3'-Processing must occur before DNA synthesis can initiate because DNA polymerases require a 3' hydroxyl to extend from.
DNA polymerases
Pol β is the main human polymerase that catalyzes short-patch BER, with pol λ able to compensate in its absence.[8] These polymerases are members of the Pol X family and typically insert only a single nucleotide. In addition to polymerase activity, these enzymes have a lyase domain that removes the 5' dRP left behind by AP endonuclease cleavage. During long-patch BER, DNA synthesis is thought to be mediated by pol δ and pol ε along with the processivity factor PCNA, the same polymerases that carry out DNA replication. These polymerases perform displacing synthesis, meaning that the downstream 5' DNA end is "displaced" to form a flap (see diagram above). Pol β can also perform long-patch displacing synthesis and can, therefore, participate in either BER pathway.[9] Long-patch synthesis typically inserts 2-10 new nucleotides.
Flap endonuclease
FEN1 removes the 5' flap generated during long patch BER. This endonuclease shows a strong preference for a long 5' flap adjacent to a 1-nt 3' flap.[10] The yeast homolog of FEN1 is RAD27. In addition to its role in long-patch BER, FEN1 cleaves flaps with a similar structure during Okazaki fragment processing, an important step in lagging strand DNA replication.
DNA ligase
DNA ligase III along with its cofactor XRCC1 catalyzes the nick-sealing step in short-patch BER in humans. DNA ligase I ligates the break in long-patch BER.
Links between BER and cancer
Defects in a variety of DNA repair pathways lead to cancer predisposition, and BER appears to follow this pattern. Deletion mutations in BER genes have shown to result in a higher mutation rate in a variety of organisms, implying that loss of BER could contribute to the development of cancer. Indeed, somatic mutations in Pol β have been found in 30% of human cancers, and some of these mutations lead to transformation when expressed in mouse cells.[11] Mutations in the DNA glycosylase MYH are also known to increase susceptibility to colon cancer.[12]
Epigenetic deficiencies in cancers
Epigenetic alterations (epimutations) in base excision repair genes have only recently begun to be evaluated in a few cancers, compared to the numerous previous studies of epimutations in genes acting in other DNA repair pathways (such as MLH1 in mismatch repair and MGMT in direct reversal).[13] Some examples of epimutations in base excision repair genes that occur in cancers are summarized below.
MBD4
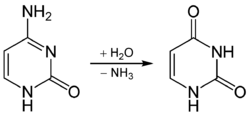
MBD4 (methyl-CpG-binding domain protein 4) is a glycosylase employed in an initial step of base excision repair. MBD4 protein binds preferentially to fully methylated CpG sites and to the altered DNA bases at those sites. These altered bases arise from the frequent hydrolysis of cytosine to uracil (see image) and hydrolysis of 5-methylcytosine to thymine, producing G:U and G:T base pairs.[14] If the improper uracils or thymines in these base pairs are not removed before DNA replication, they will cause transition mutations. MBD4 specifically catalyzes the removal of T and U paired with guanine (G) within CpG sites.[15] This is an important repair function since about 1/3 of all intragenic single base pair mutations in human cancers occur in CpG dinucleotides and are the result of G:C to A:T transitions.[15][16] These transitions comprise the most frequent mutations in human cancer. For example, nearly 50% of somatic mutations of the tumor suppressor gene p53 in colorectal cancer are G:C to A:T transitions within CpG sites.[15] Thus, a decrease in expression of MBD4 could cause an increase in carcinogenic mutations.
MBD4 expression is reduced in almost all colorectal neoplasms due to methylation of the promoter region of MBD4.[17] Also MBD4 is deficient due to mutation in about 4% of colorectal cancers.[18]
A majority of histologically normal fields surrounding neoplastic growths (adenomas and colon cancers) in the colon also show reduced MBD4 mRNA expression (a field defect) compared to histologically normal tissue from individuals who never had a colonic neoplasm.[17] This finding suggests that epigenetic silencing of MBD4 is an early step in colorectal carcinogenesis.
In a Chinese population that was evaluated, the MBD4 Glu346Lys polymorphism was associated with about a 50% reduced risk of cervical cancer, suggesting that alterations in MBD4 may be important in cancer.[19]
NEIL1
NEIL1 recognizes (targets) and removes certain oxidatively-damaged bases and then incises the abasic site via β,δ elimination, leaving 3′ and 5′ phosphate ends. NEIL1 recognizes oxidized pyrimidines, formamidopyrimidines, thymine residues oxidized at the methyl group, and both stereoisomers of thymine glycol.[20] The best substrates for human NEIL1 appear to be the hydantoin lesions, guanidinohydantoin, and spiroiminodihydantoin that are further oxidation products of 8-oxoG. NEIL1 is also capable of removing lesions from single-stranded DNA as well as from bubble and forked DNA structures. A deficiency in NEIL1 causes increased mutagenesis at the site of an 8-oxo-Gua:C pair, with most mutations being G:C to T:A transversions.[21]
A study in 2004 found that 46% of primary gastric cancers had reduced expression of NEIL1 mRNA, though the mechanism of reduction was not known.[22] This study also found that 4% of gastric cancers had mutations in NEIL1. The authors suggested that low NEIL1 activity arising from reduced expression and/or mutation in NEIL1 was often involved in gastric carcinogenesis.
A screen of 145 DNA repair genes for aberrant promoter methylation was performed on head and neck squamous cell carcinoma (HNSCC) tissues from 20 patients and from head and neck mucosa samples from 5 non-cancer patients.[23] This screen showed that NEIL1, with substantially increased hypermethylation, had the most significantly different frequency of methylation. Furthermore, the hypermethylation corresponded to a decrease in NEIL1 mRNA expression. Further work with 135 tumor and 38 normal tissues also showed that 71% of HNSCC tissue samples had elevated NEIL1 promoter methylation.[23]
When 8 DNA repair genes were evaluated in non-small cell lung cancer (NSCLC) tumors, 42% were hypermethylated in the NEIL1 promoter region.[24] This was the most frequent DNA repair abnormality found among the 8 DNA repair genes tested. NEIL1 was also one of six DNA repair genes found to be hypermethylated in their promoter regions in colorectal cancer.[25]
See also
- DNA repair
- DNA mismatch repair
- Nucleotide excision repair
- Homologous recombination
- Non-homologous end joining
References
- ↑ Liu Y, Prasad R, Beard WA, Kedar PS, Hou EW, Shock DD, Wilson SH (2007). "Coordination of Steps in Single-nucleotide Base Excision Repair Mediated by Apurinic/Apyrimidinic Endonuclease 1 and DNA Polymerase β". Journal of Biological Chemistry. 282 (18): 13532–13541. doi:10.1074/jbc.M611295200. PMC 2366199
 . PMID 17355977.
. PMID 17355977. - ↑ Jayanta Chaudhuri & Frederick W. Alt (2004). "Class-switch recombination: interplay of transcription, DNA deamination and DNA repair". Nature Reviews Immunology. 4 (7): 541–552. doi:10.1038/nri1395. PMID 15229473.
- ↑ Fortini P, Dogliotti E (April 2007). "Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways". DNA Repair. 6 (4): 398–409. doi:10.1016/j.dnarep.2006.10.008. PMID 17129767.
- ↑ Gellon L, Carson DR, Carson JP, Demple B (February 2008). "Intrinsic 5'-Deoxyribose-5-phosphate Lyase Activity in Saccharomyces cerevisiae Trf4 Protein with a Possible Role in Base Excision DNA Repair". DNA Repair. 7 (2): 187–98. doi:10.1016/j.dnarep.2007.09.009. PMC 2258243. PMID 17983848.
- ↑ Fromme JC, Banerjee A, Verdine GL (February 2004). "DNA glycosylase recognition and catalysis". Curr. Opin. Struct. Biol. 14 (1): 43–9. doi:10.1016/j.sbi.2004.01.003. PMID 15102448.
- ↑ Aravind L, Walker DR, Koonin EV (1999). "Conserved domains in DNA repair proteins and evolution of repair systems". Nucleic Acids Research. 27 (5): 1223–1242. doi:10.1093/nar/27.5.1223. PMC 148307. PMID 9973609.
- ↑ Demple B, Herman T, Chen DS (1991). "Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes". PNAS USA. 88 (24): 11450–11454. doi:10.1073/pnas.88.24.11450. PMC 53153. PMID 1722334.
- ↑ Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH (May 2005). "DNA polymerase lambda mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts". J. Biol. Chem. 280 (18): 18469–75. doi:10.1074/jbc.M411864200. PMID 15749700.
- ↑ Beard WA, Prasad R, Wilson SH (2006). "Activities and mechanism of DNA polymerase beta". Meth. Enzymol. 408: 91–107. doi:10.1016/S0076-6879(06)08007-4. PMID 16793365.
- ↑ Kao HI, Henricksen LA, Liu Y, Bambara RA (April 2002). "Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate". J. Biol. Chem. 277 (17): 14379–89. doi:10.1074/jbc.M110662200. PMID 11825897.
- ↑ Starcevic D, Dalal S, Sweasy JB (August 2004). "Is there a link between DNA polymerase beta and cancer?". Cell Cycle. 3 (8): 998–1001. doi:10.4161/cc.3.8.1062. PMID 15280658.
- ↑ Farrington, S. M.; Tenesa, A; Barnetson, R; Wiltshire, A; Prendergast, J; Porteous, M; Campbell, H; Dunlop, M. G. (2005). "Germline susceptibility to colorectal cancer due to base-excision repair gene defects". The American Journal of Human Genetics. 77 (1): 112–9. doi:10.1086/431213. PMC 1226182. PMID 15931596.
- ↑ Carol Bernstein and Harris Bernstein (2015). Epigenetic Reduction of DNA Repair in Progression to Cancer, Advances in DNA Repair, Prof. Clark Chen (Ed.), ISBN 978-953-51-2209-8, InTech, Available from: http://www.intechopen.com/books/advances-in-dna-repair/epigenetic-reduction-of-dna-repair-in-progression-to-cancer
- ↑ Bellacosa A, Drohat AC (Aug 2015). "Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites". DNA Repair. 32: 33–42. doi:10.1016/j.dnarep.2015.04.011. PMID 26021671.
- 1 2 3 Sjolund AB, Senejani AG, Sweasy JB (2013). "MBD4 and TDG: multifaceted DNA glycosylases with ever expanding biological roles". Mutation Research. 743-744: 12–25. doi:10.1016/j.mrfmmm.2012.11.001. PMC 3661743. PMID 23195996.
- ↑ Cooper DN, Youssoufian H (Feb 1988). "The CpG dinucleotide and human genetic disease". Human Genetics. 78 (2): 151–5. doi:10.1007/bf00278187. PMID 3338800.
- 1 2 Howard JH, Frolov A, Tzeng CW, Stewart A, Midzak A, Majmundar A, Godwin A, Heslin M, Bellacosa A, Arnoletti JP (Jan 2009). "Epigenetic downregulation of the DNA repair gene MED1/MBD4 in colorectal and ovarian cancer". Cancer Biology & Therapy. 8 (1): 94–100. doi:10.4161/cbt.8.1.7469. PMC 2683899. PMID 19127118.
- ↑ Tricarico R, Cortellino S, Riccio A, Jagmohan-Changur S, Van der Klift H, Wijnen J, Turner D, Ventura A, Rovella V, Percesepe A, Lucci-Cordisco E, Radice P, Bertario L, Pedroni M, Ponz de Leon M, Mancuso P, Devarajan K, Cai KQ, Klein-Szanto AJ, Neri G, Møller P, Viel A, Genuardi M, Fodde R, Bellacosa A (Oct 2015). "Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis". Oncotarget. doi:10.18632/oncotarget.5740. PMID 26503472.
- ↑ Xiong XD, Luo XP, Liu X, Jing X, Zeng LQ, Lei M, Hong XS, Chen Y (2012). "The MBD4 Glu346Lys polymorphism is associated with the risk of cervical cancer in a Chinese population". Int. J. Gynecol. Cancer. 22 (9): 1552–6. doi:10.1097/IGC.0b013e31826e22e4. PMID 23027038.
- ↑ Nemec AA, Wallace SS, Sweasy JB (Oct 2010). "Variant base excision repair proteins: contributors to genomic instability". Seminars in Cancer Biology. 20 (5): 320–8. doi:10.1016/j.semcancer.2010.10.010. PMC 3254599. PMID 20955798.
- ↑ Suzuki T, Harashima H, Kamiya H (2010). "Effects of base excision repair proteins on mutagenesis by 8-oxo-7,8-dihydroguanine (8-hydroxyguanine) paired with cytosine and adenine". DNA Repair (Amst.). 9 (5): 542–50. doi:10.1016/j.dnarep.2010.02.004. PMID 20197241.
- ↑ Shinmura K, Tao H, Goto M, Igarashi H, Taniguchi T, Maekawa M, Takezaki T, Sugimura H (2004). "Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer". Carcinogenesis. 25 (12): 2311–7. doi:10.1093/carcin/bgh267. PMID 15319300.
- 1 2 Chaisaingmongkol J, Popanda O, Warta R, Dyckhoff G, Herpel E, Geiselhart L, Claus R, Lasitschka F, Campos B, Oakes CC, Bermejo JL, Herold-Mende C, Plass C, Schmezer P (2012). "Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma". Oncogene. 31 (49): 5108–16. doi:10.1038/onc.2011.660. PMID 22286769.
- ↑ Do H, Wong NC, Murone C, John T, Solomon B, Mitchell PL, Dobrovic A (2014). "A critical re-assessment of DNA repair gene promoter methylation in non-small cell lung carcinoma". Scientific Reports. 4: 4186. doi:10.1038/srep04186. PMC 3935198. PMID 24569633.
- ↑ Farkas SA, Vymetalkova V, Vodickova L, Vodicka P, Nilsson TK (Apr 2014). "DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/β-catenin signaling pathway genes". Epigenomics. 6 (2): 179–91. doi:10.2217/epi.14.7. PMID 24811787.
External links
- Base Excision Repair at the US National Library of Medicine Medical Subject Headings (MeSH)