Ball and chain inactivation
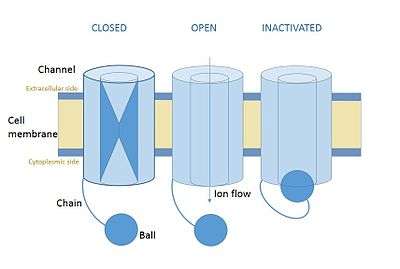
In cellular neurone science ball and chain inactivation is a model to explain the fast inactivation mechanism of voltage-gated ion channels. The process is also called hinged-lid inactivation or N-type inactivation.Is also called ball chain mechanism A voltage gated ion channel can be in three states: open, closed or inactivated. The inactive state is mainly achieved through fast inactivation, by which a channel transitions rapidly from an open to an inactivated state. The model proposes that the inactive state, which is stable and non-conducting, is caused by the physical blockage of the pore. The blockage is caused by a ball of amino acids attached to the main protein by a string of residues on the cytoplasmic side. The ball enters the open channel and binds to the hydrophobic inner vestibule at the center of the channel. The blockage causes inactivation of the channel by stopping the flow of ions.[1][2] This phenomenon has mainly been studied in potassium channels and in sodium channels, but many of the details remain unclear.[3]
Discovery and Evidence
Biochemical evidence
The initial evidence for a ball and chain inactivation came in 1977 with Clay Armstrong and Francisco Bezanilla's work.[4] The suggestion of a physical basis for non-conductance came from experiments in squid giant axons, showing that internal treatment with pronase disrupted the inactivation phenomenon. This suggested a physical, tethered mechanism for inactivation as the pronase was inferred to degrade the channel blocker and abolish the inactivation process. These experiments also showed that inactivation can only occur after the opening of the channel. This was done by hyperpolarising the membrane, causing the channel to open, and observing a delay in inactivation. Inactivation was not observed when the membrane was depolarised (closed). Introducing tetraethylammonium (TEA) on the intracellular side of the channel was found to mimic inactivation in non-inactivating channels.[5] Blockage of the channel by TEA is mutually exclusive with peptide-mediate blockage, suggesting that TEA competes for an inactivation binding site.[6]
Molecular evidence
Mutagenesis experiments have identified an intracellular string of amino acids as prime candidates for the pore blocker.[5] The precise sequence of amino acids that makes up the channel-blocking ball in potassium channels was identified through the creation a synthetic peptide. The peptide was built based on the sequence of a 20 amino acid residue from the Drosophila melanogaster 's Shaker ShB protein and applied on the intracellular side of a non-inactivating channel in Xenopus oocytes. The peptide restored inactivation to the channel, giving further support to the ball and chain model. In β2 proteins, the first three residues after the initial methionine have been identified as essential for inactivation. The initial residues have a sequence motif of phenylalanine, isoleucine and tryptophan (FIW) without which inactivation does not occur. Modifying the subsequent residues alters the speed and efficacy of inactivation without abolishing it.[7]
Structural evidence
More recently, nuclear magnetic resonance studies in Xenopus oocyte BK channels have shed further light on the structural properties of the ball and chain domain.[8] The introduction of the KCNMB2 β subunit to the cytoplasmic side of a non-inactivating channel restored inactivation, conforming to the expected behaviour of a ball and chain-type protein. NMR analysis showed that the ball domain is composed of residues 1-17 and the chain region of residues 20-45. The three amino acids in the middle constitute a flexible linker region between the two functional regions. The ball is at the N-terminus of the β subunit and consists of a disordered part (residues 1–10) and a loop-helix motif formed by a block of amino acids spanning from serine at position 11 to aspartate at position 16. The structure of the chain domain is 4-turn alpha helix structure.
Structure
The ball and chain domains are on the cytoplasmic side of the channel. The most precise structural studies have been carried out in Shaker potassium channels, in which the precise residues involved in the process have been identified. The first 19 amino acids of the N-terminus constitute the ball domain. This is made up of 11 hydrophobic amino acids, 8 hydrophilic ones and 4 positively charged ones.[9] The following 60 amino acids constitute the chain domain. Modifying the amino acids of the ball while preserving their chemical properties does not disrupt the inactivation mechanism. This suggests that the ball occludes the channel by binding electrostatically rather than covalently.[10] Structural studies have shown that the inner pore of the potassium channel is accessible only through side slits between the cytoplasmic domains of the four α-subunits, rather than from a central route as previously thought.[11] The ball domain enters the channel through the side slits and attaches to a binding site deep in the central cavity. This process involves a conformational change, which allows the ball and chain blocker to elongate and reach the inner center of the channel.[12]
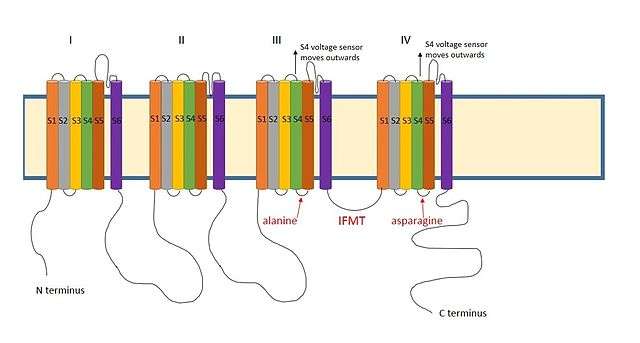
A positively charged region between the III and IV domains of sodium channels is thought to act in a similar way.[9] The essential region for inactivation in sodium channels is four amino acid sequence made up of isoleucine, phenylalanine, methionine and threonine (IFMT)[13] The T and F interact directly with the docking site in the channel pore.[14] When voltage-gated sodium channels open, the S4 segment moves outwards from the channel and into the extracellular side. This exposes hydrophobic residues in the S4 and S5 segments which interact with the inactivation ball. The phenylalanine of the ball interacts with the alanine in domain III's S4-S5 segments and the asparagine in domain IV's S4-S5 segments.[15] This explains why inactivation can only occur once the channel is open.
Lateral slits are also present in sodium channels,[16] suggesting that the access route for the ball domain may be similar.
There is a distinction between direct inactivation and two-step inactivation. Direct inactivation, which occurs in Shaker potassium channels results from the direct blockage of the channel by the ball protein, while two-step inactivation, thought to occur in BK channels, requires an intermediate binding step.[17]
Effects on neuronal firing
The interplay between opening and inactivation controls the firing pattern of a neuron by changing the rate and amount of ion flow though the channels. Voltage-gated ion channels open upon depolarization of the cell membrane. This creates a current caused by the flow of ions through the channel. Shortly after opening, the channel is blocked by the peptide ball. When the membrane repolarises, the ball is expelled from the channel. This causes a resurgent current: a flow of ions between inactivation and closure of the channel.[18] In sodium channels this process is modulated by β subunits. The β1 subunit aids recovery from inactivation,[19] while β2 accelerates inactivation.[20] The β subunits can also interfere with ball and chain domains by blocking their entry into the channel. This leads to persistent currents, caused by the continued influx of ions. The β3 subunit can increase persistent current in certain sodium channels.[13]
Implications for Disease
Differences in persistent and resurgent currents have been implicated in certain human neurological and neuromuscular disorders. In epilepsy, mutations in sodium channels genes delay inactivation. This leads to the channel staying open for longer and thus longer-lasting neuronal firing.[21] Higher levels of persistent current are observed in epilepsy. This constant, low-level neuronal stimulation has been linked to the seizures typical of this disorder.[22]
Inactivation anomalies have also been linked to Brugada syndrome. Mutations in genes encoding the α subunit in cardiac sodium channels affect inactivation. These increase persistent current by interfering with inactivation, though different mutations have opposite effects in inactivation speed.[23]
Mutations in the α subunit of skeletal muscles are also associated with myotonia. The characteristic muscular hyperexcitation of mytonia is mainly caused by the presence sodium channels which do not inactivate, causing high levels of persistent current in the muscles.[24]
Inactivation Prevention Domain
Potassium channels have an additional feature in the N-terminus which makes the channels unable to inactivate. The N-type inactivation-prevention (NIP) domain counteracts the effect of the peptide ball. Channels containing the NIP domain behave as mutated non-inactivating channels, as they have no inactivation activity.[25] The effect is thought be stoichiometric, as the gradual introduction of un-tethered synthetic balls to the cytoplasm eventually restores inactivation.[26]
References
- ↑ Nicholls JG, Martin AR, Wallace BG, Fuchs PA (2011). From neuron to brain (8th ed.). Sunderland, Mas.: Sinauer Associates. pp. 123–124. ISBN 978-0878936090.
- ↑ Brady S, Siegel G, Albers RW, Price D (2012). Basic neurochemistry: molecular, cellular and medical aspects (8th ed.). Amsterdam; London: Academic Press. pp. 106–107. ISBN 978-0080959016.
- ↑ Aldrich RW (2001). "Fifty years of inactivation". Nature. Nature Publishing Group. 411 (6838): 643–644. doi:10.1038/35079705.
- ↑ Armstrong CM & Bezanilla, F (1977). "Inactivation of the sodium channel. II. Gating current experiments.". The Journal of General Physiology. Rockefeller University Press. 70 (5): 567–590. doi:10.1085/jgp.70.5.567.
- 1 2 Zagotta WN, Hoshi T, Aldrich RW (1990). "Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB.". Science. American Association for the Advancement of Science. 250 (4980): 568–571. doi:10.1126/science.2122520.
- ↑ Choi KL, Aldrich RW, Yellen G (1991). "Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels.". Proceedings of the National Academy of Sciences. National Academy of Science. 88 (12): 5092–5095. doi:10.1073/pnas.88.12.5092.
- ↑ Xia XM, Ding JP, Lingle CJ (2003). "Inactivation of BK Channels by the NH2 Terminus of the beta Auxiliary Subunit An Essential Role of a Terminal Peptide Segment of Three Hydrophobic Residues". The Journal of General Physiology. Rockefeller University Press. 121 (2): 125–148. doi:10.1085/jgp.20028667. PMC 2217327
 . PMID 12566540.
. PMID 12566540. - ↑ Bentrop D, Beyermann M, Wissmann R, Fakler B (2001). "NMR structure of the "ball-and-chain" domain of KCNMB2, the beta2-subunit of large conductance Ca2+-and voltage-activated potassium channels". Journal of Biological Chemistry. American Society for Biochemistry and Molecular Biology. 276 (45): 42116–42121. doi:10.1074/jbc.M107118200. PMID 11517232.
- 1 2 Hall ZW (1992). An introduction to molecular neurobiology (1st ed.). Sunderland, Mas.: Sinauer Associates. p. 113. ISBN 978-0878933075.
- ↑ Holmgren M, Jurman ME, Yellen G (1996). "N-type inactivation and the S4-S5 region of the Shaker K+ channel". The Journal of General Physiology. Rockefeller University Press. 108 (3): 195–206. doi:10.1085/jgp.108.3.195.
- ↑ Sokolova O, Kolmakova-Partensky L, Grigorieff N (2001). "Three-dimensional structure of a voltage-gated potassium channel at 2.5 nm resolution". Structure. Elsevier. 9 (3): 215–220. doi:10.1016/s0969-2126(01)00578-0.
- ↑ Zhou M, Morais-Cabral JH, Mann S, MacKinnon R (2002). "Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors". Nature. Nature Publishing Group. 411 (6838): 657–661. doi:10.1038/35079500.
- 1 2 3 Goldin AL (2003). "Mechanisms of sodium channel inactivation". Current Opinion in Neurobiology. Elsevier. 13 (3): 284–290. doi:10.1016/S0959-4388(03)00065-5.
- ↑ Miyamoto K, Nakagawa T, Kuroda Y (2001). "Solution structure of the cytoplasmic linker between domain III-S6 and domain IV-S1 (III--IV linker) of the rat brain sodium channel in SDS micelles". Biopolymers. Wiley Online Library. 59 (5): 380–393. doi:10.1002/1097-0282(20011015)59:5<380::AID-BIP1035>3.0.CO;2-T.
- ↑ Miyamoto K, Nakagawa T, Kuroda Y (2001). "Solution structures of the cytoplasmic linkers between segments S4 and S5 (S4- S5) in domains III and IV of human brain sodium channels in SDS micelles". The Journal of Peptide Research. Wiley Online Library. 58 (3): 193–203. doi:10.1034/j.1399-3011.2001.00912.x.
- ↑ Payandeh J, Scheuer T, Zheng N, Catterall WA (2011). "The crystal structure of a voltage-gated sodium channel". Nature. Nature Publishing Group. 475 (7356): 353–358. doi:10.1038/nature10238.
- ↑ Gonzalez-Perez V, Zeng XH, Henzler-Wildman K, Lingle CJ (2012). "Stereospecific binding of a disordered peptide segment mediates BK channel inactivation". Nature. Nature Publishing Group. 485 (7396): 133–136. doi:10.1038/nature10994.
- ↑ Bant JS, Raman IM (2010). "Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons". Proceedings of the National Academy of Sciences. National Academy of Sciences. 107 (27): 12357–12362. doi:10.1073/pnas.1005633107.
- ↑ Zimmer T & Benndorf K (2002). "The human heart and rat brain IIA Na+ channels interact with different molecular regions of the beta subunit". The Journal of General Physiology. Rockefeller University Press. 120 (6): 887–895. doi:10.1085/jgp.20028703.
- ↑ McCormick KA, Isom LL, Ragsdale D, Smith D, Scheuer T, Catterall WA (1998). "Molecular determinants of Na+ channel function in the extracellular domain of the beta1 subunit". Journal of Biological Chemistry. American Society for Biochemistry and Molecular Biology. 273 (7): 3954–3962. doi:10.1074/jbc.273.7.3954.
- ↑ Alekov AK, Rahman MM, Mitrovic N, Lehmann-Horn F, Lerche H (2000). "A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro". The Journal of Physiology. Wiley Online Library. 529 (3): 533–540. doi:10.1111/j.1469-7793.2000.00533.x.
- ↑ Stafstrom CE (2007). "Persistent sodium current and its role in epilepsy". Epilepsy Currents. Wiley Online Library. 7 (1): 15–22. doi:10.1111/j.1535-7511.2007.00156.x.
- ↑ Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, Napolitano C, Priori SG, Kass RS (2001). "Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes". Journal of Biological Chemistry. American Society for Biochemistry and Molecular Biology. 276 (33): 30623–30630. doi:10.1074/jbc.M104471200.
- ↑ Lerche H, Heine R, Pika U, George AL, Mitrovic N, Browatzki M, Weiss T, Rivet-Bastide M, Franke C, Lomonaco M (1993). "Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker". The Journal of Physiology. Wiley Online Library. 470 (1): 113–120. doi:10.1113/jphysiol.1993.sp019843. PMC 1143902. PMID 8308722.
- ↑ Roeper J, Sewing S, Zhang Y, Sommer T, Wanner SG, Pongs O (1998). "NIP domain prevents N-type inactivation in voltage-gated potassium channels". Nature. Nature Publishing Group. 391 (6665): 390–393. doi:10.1038/34916.
- ↑ Yellen G (1998). "The moving parts of voltage-gated ion channels". Quarterly Reviews of Biophysics. Cambridge University Press. 31 (3): 239–295. doi:10.1017/s0033583598003448.