Pharmacogenomics
| Part of a series on |
| Genetics |
|---|
 |
| Key components |
| History and topics |
| Research |
|
| Personalized medicine |
| Personalized medicine |
|
Pharmacogenomics is the study of the role of the genome in drug response. Its name (pharmaco- + genomics) reflects its combining of pharmacology and genomics. Pharmacogenomics can be defined as the technology that analyzes how the genetic makeup of an individual affects his/her response to drugs. [1] It deals with the influence of acquired and inherited genetic variation on drug response in patients by correlating gene expression or single-nucleotide polymorphisms with pharmacokinetics and pharmacodynamics (drug absorption, distribution, metabolism, and elimination), as well as drug receptor target effects.[2][3][4] The term pharmacogenomics is often used interchangeably with pharmacogenetics. Although both terms relate to drug response based on genetic influences, pharmacogenetics focuses on single drug-gene interactions, while pharmacogenomics encompasses a more genome-wide association approach, incorporating genomics and epigenetics while dealing with the effects of multiple genes on drug response.[5][6][7]
Pharmacogenomics aims to develop rational means to optimize drug therapy, with respect to the patients' genotype, to ensure maximum efficacy with minimal adverse effects.[8] Through the utilization of pharmacogenomics, it is hoped that pharmaceutical drug treatments can deviate from what is dubbed as the "one-dose-fits-all" approach. It attempts to eliminate the trial-and-error method of prescribing, allowing physicians to take into consideration their patient's genes, the functionality of these genes, and how this may affect the efficacy of the patient's current or future treatments (and where applicable, provide an explanation for the failure of past treatments).[5] Such approaches promise the advent of precision medicine and even personalized medicine, in which drugs and drug combinations are optimized for narrow subsets of patients or even for each individual's unique genetic makeup.[9][10] Whether used to explain a patient's response or lack thereof to a treatment, or act as a predictive tool, it hopes to achieve better treatment outcomes, greater efficacy, minimization of the occurrence of drug toxicities and adverse drug reactions (ADRs). For patients who have lack of therapeutic response to a treatment, alternative therapies can be prescribed that would best suit their requirements. In order to provide pharmacogenomic recommendations for a given drug, two possible types of input can be used: genotyping or exome or whole genome sequencing.[11] Sequencing provides many more data points, including detection of mutations that prematurely terminate the synthesized protein (early stop codon).[11]
History
Pharmacogenomics was first recognized by Pythagoras around 510 BC when he made a connection between the dangers of fava bean ingestion with hemolytic anemia and oxidative stress. Interestingly, this identification was later validated and attributed to deficiency of G6PD in the 1950s and called favism.[12][13] Although the first official publication dates back to 1961,[14] circa 1950s marked the unofficial beginnings of this science. Reports of prolonged paralysis and fatal reactions linked to genetic variants in patients who lacked butyryl-cholinesterase (‘pseudocholinesterase’) following administration of succinylcholine injection during anesthesia were first reported in 1956.[2][15] The term pharmacogenetic was first coined in 1959 by Friedrich Vogel of Heidelberg, Germany (although some papers suggest it was 1957). In the late 1960s, twin studies supported the inference of genetic involvement in drug metabolism, with identical twins sharing remarkable similarities to drug response compared to fraternity twins.[16] The term pharmacogenomics first began appearing around the 1990s.[12]
The first FDA approval of a pharmacogenetic test was in 2005[17] (for alleles in CYP2D6 and CYP2C19).
Drug-metabolizing enzymes
There are several known genes which are largely responsible for variances in drug metabolism and response. The focus of this article will remain on the genes that are more widely accepted and utilized clinically for brevity.
- Cytochrome P450s
- VKORC1
- TPMT
Cytochrome P450
The most prevalent drug-metabolizing enzymes (DME) are the Cytochrome P450 (CYP) enzymes. The term Cytochrome P450 was coined by Omura and Sato in 1962 to describe the membrane-bound, heme-containing protein characterized by 450 nm spectral peak when complexed with carbon monoxide.[18] The human CYP family consists of 57 genes, with 18 families and 44 subfamilies. CYP proteins are conveniently arranged into these families and subfamilies on the basis of similarities identified between the amino acid sequences. Enzymes that share 35-40% identity are assigned to the same family by an Arabic numeral, and those that share 55-70% make up a particular subfamily with a designated letter.[19] For example, CYP2D6 refers to family 2, subfamily D, and gene number 6.
From a clinical perspective, the most commonly tested CYPs include: CYP2D6, CYP2C19, CYP2C9, CYP3A4 and CYP3A5. These genes account for the metabolism of approximately 80-90% of currently available prescription drugs.[20][21] The table below provides a summary for some of the medications that take these pathways.
| Drug Metabolism of Major CYPs[22][23] | |||||
|---|---|---|---|---|---|
| Enzyme | Fraction of drug metabolism (%) | Example Drugs | |||
| CYP2C9 | 10 | Tolbutamide, ibuprofen, mefenamic acid, tetrahydrocannabinol, losartan, diclofenac | |||
| CYP2C19 | 5 | S-mephenytoin, amitriptyline, diazepam, omeprazole, proguanil, hexobarbital, propranolol, imipramine | |||
| CYP2D6 | 20-30 | Debrisoquine, metoprolol, sparteine, propranolol, encainide, codeine, dextromethorphan, clozapine, desipramine, haloperidol, amitriptyline, imipramine | |||
| CYP3A4 | 40-45 | Erythromycin, ethinyl estradiol, nifedipine, triazolam, cyclosporine, amitriptyline, imipramine | |||
| CYP3A5 | <1 | Erythromycin, ethinyl estradiol, nifedipine, triazolam, cyclosporine, amitriptyline, aldosterone | |||
CYP2D6
Also known as debrisoquine hydroxylase (named after the drug that led to its discovery), CYP2D6 is the most well-known and extensively studied CYP gene.[24] It is a gene of great interest also due to its highly polymorphic nature, and involvement in a high number of medication metabolisms (both as a major and minor pathway). More than 100 CYP2D6 genetic variants have been identified.[23]
CYP2C19
Discovered in the early 1980s, CYP2C19 is the second most extensively studied and well understood gene in pharmacogenomics.[22] Over 28 genetic variants have been identified for CYP2C19,[25] of which affects the metabolism of several classes of drugs, such as antidepressants and proton pump inhibitors.[26]
CYP2C9
CYP2C9 constitutes the majority of the CYP2C subfamily, representing approximately 20% of the liver content. It is involved in the metabolism of approximately 10% of all drugs, which include medications with narrow therapeutic windows such as warfarin and tolbutamide.[26][27] There are approximately 57 genetic variants associated with CYP2C9.[25]
CYP3A4 and CYP3A5
The CYP3A family is the most abundantly found in the liver, with CYP3A4 accounting for 29% of the liver content.[22] These enzymes also cover between 40-50% of the current prescription drugs, with the CYP3A4 accounting for 40-45% of these medications.[13] CYP3A5 has over 11 genetic variants identified at the time of this publication.[25]
VKORC1
The vitamin K epoxide reductase complex subunit 1 (VKORC1) is responsible for the pharmacodynamics of warfarin.[28] VKORC1 along with CYP2C9 are useful for identifying the risk of bleeding during warfarin administration. Warfarin works by inhibiting VKOR, which is encoded by the VKORC1 gene. Individuals with polymorphism in this have an affected response to warfarin treatment.[29]
TPMT
Thiopurine methyltransferase (TPMT) catalyzes the S-methylation of thiopurines, thereby regulating the balance between cytotoxic thioguanine nucleotide and inactive metabolites in hematopoietic cells.[30] TPMT is highly involved in 6-MP metabolism and TMPT activity and TPMT genotype is known to affect the risk of toxicity. Excessive levels of 6-MP can cause myelosuppression and myelotoxicity.[31]
Codeine, clopidogrel, tamoxifen, and warfarin a few examples of medications that follow the above metabolic pathways.
Predictive prescribing
Patient genotypes are usually categorized into the following predicted phenotypes:
- Ultra-Rapid Metabolizer: Patients with substantially increased metabolic activity.
- Extensive Metabolizer: Normal metabolic activity;
- Intermediate Metabolizer: Patients with reduced metabolic activity; and
- Poor Metabolizer: Patients with little to no functional metabolic activity.
The two extremes of this spectrum are the Poor Metabolizers and Ultra-Rapid Metabolizers. Efficacy of a medication is not only based on the above metabolic statuses, but also the type of drug consumed. Drugs can be classified into two main groups: active drugs and prodrugs. Active drugs refer to drugs that are inactivated during metabolism, and prodrugs are inactive until they are metabolized.
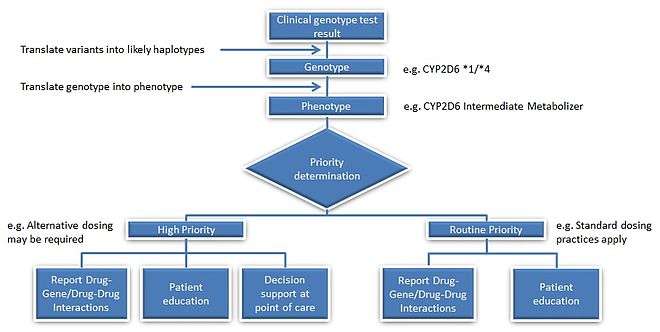
For example, we have two patients who are taking codeine for pain relief. Codeine is a prodrug, so it requires conversion from its inactive form to its active form. The active form of codeine is morphine, which provides the therapeutic effect of pain relief. If person A receives one *1 allele each from mother and father to code for the CYP2D6 gene, then that person is considered to have an extensive metabolizer (EM) phenotype, as allele *1 is considered to have a normal-function (this would be represented as CYP2D6 *1/*1). If person B on the other hand had received one *1 allele from the mother and a *4 allele from the father, that individual would be an Intermediate Metabolizer (IM) (the genotype would be CYP2D6 *1/*4). Although both individuals are taking the same dose of codeine, person B could potentially lack the therapeutic benefits of codeine due to the decreased conversion rate of codeine to its active counterpart morphine.
Each phenotype is based upon the allelic variation within the individual genotype. However, several genetic events can influence a same phenotypic trait, and establishing genotype-to-phenotype relationships can thus be far from consensual with many enzymatic patterns. For instance, the influence of the CYP2D6*1/*4 allelic variant on the clinical outcome in patients treated with Tamoxifen remains debated today. In oncology, genes coding for DPD, UGT1A1, TPMT, CDA involved in the pharmacokinetics of 5-FU/capecitabine, irinotecan, 6-mercaptopurine and gemcitabine/cytarabine, respectively, have all been described as being highly polymorphic. A strong body of evidence suggests that patients affected by these genetic polymorphisms will experience severe/lethal toxicities upon drug intake, and that pre-therapeutic screening does help to reduce the risk of treatment-related toxicities through adaptive dosing strategies.[32]
Applications
The list below provides a few more commonly known applications of pharmacogenomics:[33]
- Improve drug safety, and reduce ADRs;
- Tailor treatments to meet patients' unique genetic pre-disposition, identifying optimal dosing;
- Improve drug discovery targeted to human disease; and
- Improve proof of principle for efficacy trials.
Pharmacogenomics may be applied to several areas of medicine, including Pain Management, Cardiology, Oncology, and Psychiatry. A place may also exist in Forensic Pathology, in which pharmacogenomics can be used to determine the cause of death in drug-related deaths where no findings emerge using autopsy.[34]
In cancer treatment, pharmacogenomics tests are used to identify which patients are most likely to respond to certain cancer drugs. In behavioral health, pharmacogenomic tests provide tools for physicians and care givers to better manage medication selection and side effect amelioration. Pharmacogenomics is also known as companion diagnostics, meaning tests being bundled with drugs. Examples include KRAS test with cetuximab and EGFR test with gefitinib. Beside efficacy, germline pharmacogenetics can help to identify patients likely to undergo severe toxicities when given cytotoxics showing impaired detoxification in relation with genetic polymorphism, such as canonical 5-FU.[35]
In cardiovascular disorders, the main concern is response to drugs including warfarin, clopidogrel, beta blockers, and statins.[11]
Example case studies
Case A – Antipsychotic adverse reaction[36]
Patient A suffers from schizophrenia. Their treatment included a combination of ziprasidone, olanzapine, trazodone and benzotropine. The patient experienced dizziness and sedation, so they were tapered off ziprasidone and olanzapine, and transition to quetiapine. Trazodone was discontinued. The patient then experienced excessive sweating, tachycardia and neck pain, gained considerable weight and had hallucinations. Five months later, quetiapine was tapered and discontinued, with ziprasidone re-introduction into their treatment due to the excessive weight gain. Although the patient lost the excessive weight they gained, they then developed muscle stiffness, cogwheeling, tremor and night sweats. When benztropine was added they experienced blurry vision. After an additional five months, the patient was switched from ziprasidone to aripiprazole. Over the course of 8 months, patient A gradually experienced more weight gain, sedation, developed difficulty with their gait, stiffness, cogwheel and dyskinetic ocular movements. A pharmacogenomics test later proved the patient had a CYP2D6 *1/*41, with has a predicted phenotype of IM and CYP2C19 *1/*2 with predicted phenotype of IM as well.
Case B – Pain Management [37]
Patient B is a woman who gave birth by caesarian section. Her physician prescribed codeine for post-caesarian pain. She took the standard prescribed dose, however experienced nausea and dizziness while she was taking codeine. She also noticed that her breastfed infant was lethargic and feeding poorly. When the patient mentioned these symptoms to her physician, they recommended that she discontinue codeine use. Within a few days, both the patient and her infant’s symptoms were no longer present. It is assumed that if the patient underwent a pharmacogenomic test, it would have revealed she may have had a duplication of the gene CYP2D6 placing her in the Ultra-rapid metabolizer (UM) category, explaining her ADRs to codeine use.
Case C – FDA Warning on Codeine Overdose for Infants[38]
On February 20, 2013, the FDA released a statement addressing a serious concern regarding the connection between children who are known as CYP2D6 UM and fatal reactions to codeine following tonsillectomy and/or adenoidectomy (surgery to remove the tonsils and/or adenoids). They released their strongest Boxed Warning to elucidate the dangers of CYP2D6 UMs consuming codeine. Codeine is converted to morphine by CYP2D6, and those who have UM phenotypes are at danger of producing large amounts of morphine due to the increased function of the gene. The morphine can elevate to life-threatening or fatal amounts, as became evident with the death of three children in August 2012.
Polypharmacy
A potential role pharmacogenomics may play would be to reduce the occurrence of polypharmacy. It is theorized that with tailored drug treatments, patients will not have the need to take several medications that are intended to treat the same condition. In doing so, they could potentially minimize the occurrence of ADRs, have improved treatment outcomes, and can save costs by avoiding purchasing extraneous medications. An example of this can be found in Psychiatry, where patients tend to be receiving more medications than even age-matched non-psychiatric patients. This has been associated with an increased risk of inappropriate prescribing.[39]
The need for pharmacogenomics tailored drug therapies may be most evident in a survey conducted by the Slone Epidemiology Center at Boston University from February 1998 to April 2007. The study elucidated that an average of 82% of adults in the United States are taking at least one medication (prescription or nonprescription drug, vitamin/mineral, herbal/natural supplement), and 29% are taking five or more. The study suggested that those aged 65 years or older continue to be the biggest consumers of medications, with 17-19 % in this age group taking at least ten medications in a given week. Polypharmacy has also shown to have increased since 2000 from 23% to 29%.[40]
Drug labeling
The U.S. Food and Drug Administration (FDA) appears to be very invested in the science of pharmacogenomics[41] as is demonstrated through the 120 and more FDA-approved drugs that include pharmacogenomic biomarkers in their labels.[42] On May 22, 2005, the FDA issued its first Guidance for Industry: Pharmacogenomic Data Submissions, which clarified the type of pharmacogenomic data required to be submitted to the FDA and when.[43] Experts recognized the importance of the FDA’s acknowledgement that pharmacogenomics experiments will not bring negative regulatory consequences.[44] The FDA had released its latest guide Clinical Pharmacogenomics (PGx): Premarket Evaluation in Early-Phase Clinical Studies and Recommendations for Labeling in January, 2013. The guide is intended to address the use of genomic information during drug development and regulatory review processes.
Challenges
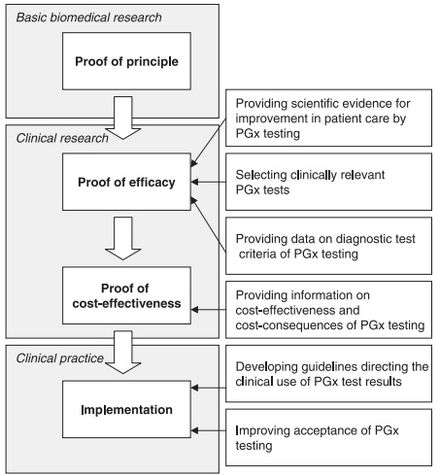
Although there appears to be a general acceptance of the basic tenet of pharmacogenomics amongst physicians and healthcare professionals,[46] several challenges exist that slow the uptake, implementation, and standardization of pharmacogenomics. Some of the concerns raised by physicians include:[7][46][47]
- Limitation on how to apply the test into clinical practices and treatment;
- A general feeling of lack of availability of the test;
- The understanding and interpretation of evidence-based research; and
- Ethical, legal and social issues.
Issues surrounding the availability of the test include:[45]
- The lack of availability of scientific data: Although there are considerable number of DME involved in the metabolic pathways of drugs, only a fraction have sufficient scientific data to validate their use within a clinical setting; and
- Demonstrating the cost-effectiveness of pharmacogenomics: Publications for the pharmacoeconomics of pharmacogenomics are scarce, therefore sufficient evidence does not at this time exist to validate the cost-effectiveness and cost-consequences of the test.
Although other factors contribute to the slow progression of pharmacogenomics (such as developing guidelines for clinical use), the above factors appear to be the most prevalent.
Controversies
Some alleles that vary in frequency between specific populations have been shown to be associated with differential responses to specific drugs. The beta blocker atenolol is an anti-hypertensive medication that is shown to more significantly lower the blood pressure of Caucasian patients than African American patients in the United States. This observation suggests that Caucasian and African American populations have different alleles governing oleic acid biochemistry, which react differentially with atenolol.[48] Similarly, hypersensitivity to the antiretroviral drug abacavir is strongly associated with a single-nucleotide polymorphism that varies in frequency between populations.[49]
The FDA approval of the drug BiDil (isosorbide dinitrate/hydralazine) with a label specifying African-Americans with congestive heart failure, produced a storm of controversy over race-based medicine and fears of genetic stereotyping,[50] even though the label for BiDil did not specify any genetic variants but was based on racial self-identification.[51][52]
Future
Computational advances in pharmacogenomics has proven to be a blessing in research. As a simple example, for nearly a decade the ability to store more information on a hard drive has enabled us to investigate a human genome sequence cheaper and in more detail with regards to the effects/risks/safety concerns of drugs and other such substances. Such computational advances are expected to continue in the future.[53] The aim is to use the genome sequence data to effectively make decisions in order to minimise the negative impacts on, say, a patient or the health industry in general. A large amount of research in the biomedical sciences regarding Pharmacogenomics as of late stems from combinatorial chemistry,[54] genomic mining, omic technologies and high throughput screening. In order for the field to grow, rich knowledge enterprises and business must work more closely together and adopt simulation strategies. Consequently, more importance must be placed on the role of computational biology with regards to safety and risk assessments. Here, we can find the growing need and importance of being able to manage large, complex data sets, being able to extract information by integrating disparate data so that developments can be made in improving human health.
Web-based resources
| Web Resources for Pharmacogenomics[55][56] | |||||
|---|---|---|---|---|---|
| Data Source | Main Use | URL | |||
| Cytochrome P450 (CYP) Allele Nomenclature Database | A comprehensive list of genes and SNPs identified in the area of pharmacogenomics | http://www.cypalleles.ki.se/ | |||
| SuperCYP Bioinformatics Tool | Containing 1170 drugs with more than 3800 interactions, and approximately 2000 known SNPs. These SNPs are listed and ordered according to their effect on expression and/or activity | http://bioinformatics.charite.de/supercyp/ | |||
| PharmGKB | The Pharmacogenomics Knowledge Base (PharmGKB) is an interactive tool for researchers investigating how genetic variation affects drug response | https://www.pharmgkb.org/ | |||
| dbSNP database | A repository of SNPs and other variants that have been reported after discovery, compiled and officially named. These are SNPs across the board | http://www.ncbi.nlm.nih.gov/SNP/ | |||
| FINDbase | Repository of allele frequencies of pharmacogenetic markers in different populations | http://www.findbase.org/ | |||
| Pharmacogenomics Biomarkers in Drug Labelling | A table that identifies which FDA-approved drugs have pharmacogenomics-related warning labels | http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm | |||
| SNPedia | A wiki-based bioinformatics database of SNPs | http://www.snpedia.com/index.php/SNPedia | |||
| Pharmacogenomics Research Network (PGRN) | The PGRN hosts resources and information to stimulate collaborative research in pharmacogenomics and precision medicine. | http://www.pgrn.org/ | |||
See also
References
- ↑ Ermak, Gennady (2015). Emerging Medical Technologies. World Scientific. ISBN 978-981-4675-80-2.
- 1 2 Johnson JA (November 2003). "Pharmacogenetics: potential for individualized drug therapy through genetics.". Trends Genet. 19 (11): 660–6. doi:10.1016/j.tig.2003.09.008. PMID 14585618.
- ↑ "Center for Pharmacogenomics and Individualized Therapy". Retrieved 2014-06-25.
- ↑ "overview of pharmacogenomics". Up-to-Date. May 16, 2014. Retrieved 2014-06-25.
- 1 2 Sheffield LJ, Phillimore HE (2009). "Clinical use of pharmacogenomic tests in 2009". Clin Biochem Rev. 30 (2): 55–65. PMC 2702214
 . PMID 19565025.
. PMID 19565025. - ↑ Shin J, Kayser SR, Langaee TY (April 2009). "Pharmacogenetics: from discovery to patient care.". Am J Health Syst Pharm. 66 (7): 625–37. doi:10.2146/ajhp080170. PMID 19299369.
- 1 2 "Center for Genetics Education".
- ↑ Becquemont L (June 2009). "Pharmacogenomics of adverse drug reactions: practical applications and perspectives". Pharmacogenomics. 10 (6): 961–9. doi:10.2217/pgs.09.37. PMID 19530963.
- ↑ "Guidance for Industry Pharmacogenomic Data Submissions" (PDF). U.S. Food and Drug Administration. March 2005. Retrieved 2008-08-27.
- ↑ Squassina A, Manchia M, Manolopoulos VG, Artac M, Lappa-Manakou C, Karkabouna S, Mitropoulos K, Del Zompo M, Patrinos GP (August 2010). "Realities and expectations of pharmacogenomics and personalized medicine: impact of translating genetic knowledge into clinical practice". Pharmacogenomics. 11 (8): 1149–67. doi:10.2217/pgs.10.97. PMID 20712531.
- 1 2 3 Huser V, Cimino JJ (2013). "Providing pharmacogenomics clinical decision support using whole genome sequencing data as input". AMIA Summits on Translational Science Proceedings. 2013: 81. PMID 24303303.
- 1 2 Pirmohamed M (2001). "Pharmacogenetics and pharmacogenomics". Br J Clin Pharmacol. 52 (4): 345–7. doi:10.1046/j.0306-5251.2001.01498.x. PMC 2014592. PMID 11678777.
- 1 2 Prasad K (2009). "Role of regulatory agencies in translating pharmacogenetics to the clinics". Clin Cases Miner Bone Metab. 6 (1): 29–34. PMC 2781218. PMID 22461095.
- ↑ Evans DA, Clarke CA (1961). "Pharmacogenetics". Br Med Bull. 17: 234–40. PMID 13697554.
- ↑ Kalow W (2006). "Pharmacogenetics and pharmacogenomics: origin, status, and the hope for personalized medicine". Pharmacogenomics J. 6 (3): 162–5. doi:10.1038/sj.tpj.6500361. PMID 16415920.
- ↑ Motulsky AG, Qi M (2006). "Pharmacogenetics, pharmacogenomics and ecogenetics". J Zhejiang Univ Sci B. 7 (2): 169–70. doi:10.1631/jzus.2006.B0169. PMC 1363768. PMID 16421980.
- ↑ Realities and Expectations of Pharmacogenomics and Personalized Medicine: Impact of Translating Genetic Knowledge into Clinical Practice. 2010
- ↑ Debose-Boyd RA (February 2007). "A helping hand for cytochrome p450 enzymes". Cell Metab. 5 (2): 81–3. doi:10.1016/j.cmet.2007.01.007. PMID 17276348.
- ↑ Nebert DW, Russell DW (October 12, 2002). "Clinical importance of the cytochromes P450". Lancet. 360 (9340): 1155–62. doi:10.1016/s0140-6736(02)11203-7. PMID 12387968.
- ↑ Hart SN, Wang S, Nakamoto K, Wesselman C, Li Y, Zhong XB (January 2008). "Genetic polymorphisms in cytochrome P450 oxidoreductase influence microsomal P450-catalyzed drug metabolism". Pharmacogenet Genomics. 18 (1): 11–24. doi:10.1097/FPC.0b013e3282f2f121. PMID 18216718.
- ↑ Gomes AM, Winter S, Klein K, Turpeinen M, Schaeffeler E, Schwab M, Zanger UM (April 2009). "Pharmacogenomics of human liver cytochrome P450 oxidoreductase: multifactorial analysis and impact on microsomal drug oxidation". Pharmacogenomics. 10 (4): 579–99. doi:10.2217/pgs.09.7. PMID 19374516.
- 1 2 3 Hasler JA (February 1999). "Pharmacogenetics of cytochromes P450". Mol Aspects Med. 20 (1-2): 25–137. PMID 10575648.
- 1 2 Ingelman-Sundberg M (April 2004). "Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future". Trends Pharmacol Sci. 25 (4): 193–200. doi:10.1016/j.tips.2004.02.007. PMID 15063083.
- ↑ Badyal DK, Dadhich AP (October 2001). "Cytochrome P450 and drug interactions" (PDF). Indian Journal of Pharmacology. 33: 248–259.
- 1 2 3 Ingelman-Sundberg, M; Nebert, DW; Sim, SC. "The Human Cytochrome P450 (CYP) Allele Nomenclature Database". Retrieved 2014-09-03.
- 1 2 Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C (December 2007). "Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects". Pharmacol Ther. 116 (3): 496–526. doi:10.1016/j.pharmthera.2007.09.004. PMID 18001838.
- ↑ Sikka R, Magauran B, Ulrich A, Shannon M (December 2005). "Bench to bedside: Pharmacogenomics, adverse drug interactions, and the cytochrome P450 system". Acad Emerg Med. 12 (12): 1227–35. doi:10.1111/j.1553-2712.2005.tb01503.x. PMID 16282513.
- ↑ Teh LK, Langmia IM, Fazleen Haslinda MH, Ngow HA, Roziah MJ, Harun R, Zakaria ZA, Salleh MZ (April 2012). "Clinical relevance of VKORC1 (G-1639A and C1173T) and CYP2C9*3 among patients on warfarin". J Clin Pharm Ther. 37 (2): 232–6. doi:10.1111/j.1365-2710.2011.01262.x. PMID 21507031.
- ↑ U.S. Food and Drug Administration (FDA). "Table of Pharmacogenomic Biomarkers in Drug Labels.". Retrieved 2014-09-03.
- 1 2 Crews KR, Hicks JK, Pui CH, Relling MV, Evans WE (October 2012). "Pharmacogenomics and individualized medicine: translating science into practice". Clin Pharmacol Ther. 92 (4): 467–75. doi:10.1038/clpt.2012.120. PMID 22948889.
- ↑ Sim SC, Kacevska M, Ingelman-Sundberg M (February 2013). "Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects". Pharmacogenomics. 13 (1): 1–11. doi:10.1038/tpj.2012.45. PMID 23089672.
- ↑ Lee SY, McLeod HL (January 2011). "Pharmacogenetic tests in cancer chemotherapy: what physicians should know for clinical application". J Pathol. 223 (1): 15–27. doi:10.1002/path.2766. PMID 20818641.
- ↑ Cohen, Nadine (November 2008). Pharmacogenomics and Personalized Medicine (Methods in Pharmacology and Toxicology). Totowa, NJ: Humana Press. p. 6. ISBN 978-1934115046.
- ↑ Pelotti, Susan; Bini, Carla (September 12, 2011). Forensic Pharmacogenetics (Forensic Medicine - From Old Problems to New Challenges). INTECH Open Access Publisher. p. 268. ISBN 978-953-307-262-3. Retrieved 2014-09-03.
- ↑ Ciccolini J, Gross E, Dahan L, Lacarelle B, Mercier C (October 2010). "Routine dihydropyrimidine dehydrogenase testing for anticipating 5-fluorouracil-related severe toxicities: hype or hope?". Clin Colorectal Cancer. 9 (4): 224–8. doi:10.3816/CCC.2010.n.033. PMID 20920994.
- ↑ Foster A, Wang Z, Usman M, Stirewalt E, Buckley P (December 2007). "Pharmacogenetics of antipsychotic adverse effects: Case studies and a literature review for clinicians". Neuropsychiatr Dis Treat. 3 (6): 965–973. PMC 2656342. PMID 19300635.
- ↑ "Pharmacogenetics: increasing the safety and effectiveness of drug therapy [Brochure]" (PDF). American Medical Association. 2011.
- ↑ "FDA Drug Safety Communication: Safety review update of codeine use in children; new Boxed Warning and Contraindication on use after tonsillectomy and/or adenoidectomy". United States Food and Drug Administration. 2013-02-20.
- ↑ Ritsner, Michael (2013). Polypharmacy in Psychiatry Practice, Volume I. Multiple Medication Strategies. Dordrecht: Springer Science and Business Media. ISBN 978-94-007-5804-9.
- ↑ "Patterns of Medication Use in the United States". Boston University, Slone Epidemiology Center. 2006.
- ↑ "Pharmacogenetics and Pharmacogenomics: State-of-the-art and potential socio-economic impacts in the EU". European Commission, Joint Research Centre, Institute for Prospective Technological Studies. 2006-04-01.
- ↑ "Table of Pharmacogenomic Biomarkers in Drug Labels". United States Food and Drug Administration. 2013-06-19.
- ↑ Xie HG, Frueh FW (2005). "Pharmacogenomics steps toward personalized medicine." (PDF). Personalized Medicine. 2 (4): 325–337. doi:10.2217/17410541.2.4.325.
- ↑ Katsnelson A (2005). "Cautious welcome for FDA pharmacogenomics guidance". Nat Biotechnol. 23 (5): 510. doi:10.1038/nbt0505-510. PMID 15877053.
- 1 2 Swen JJ, Huizinga TW, Gelderblom H, de Vries EG, Assendelft WJ, Kirchheiner J, Guchelaar HK. "Translating Pharmacogenomics: Challenges on the Road to the Clinic". PLoS Medicine. 4 (8): 1317–24. doi:10.1371/journal.pmed.0040209.
- 1 2 Stanek EJ, Sanders CL, Taber KA, Khalid M, Patel A, Verbrugge RR, Agatep BC, Aubert RE, Epstein RS, Frueh FW (Mar 2012). "Adoption of pharmacogenomic testing by US physicians: results of a nationwide survey.". Clin Pharmacol Ther. 91 (3): 450–8. doi:10.1038/clpt.2011.306. PMID 22278335.
- ↑ Ma JD, Lee KC, Kuo GM (August 2012). "Clinical application of pharmacogenomics". J Pharm Pract. 25 (4): 417–27. doi:10.1177/0897190012448309. PMID 22689709.
- ↑ Wikoff WR, Frye RF, Zhu H, Gong Y, Boyle S, Churchill E, Cooper-Dehoff RM, Beitelshees AL, Chapman AB, Fiehn O, Johnson JA, Kaddurah-Daouk R (2013). "Pharmacometabolomics reveals racial differences in response to atenolol treatment". PLoS ONE. 8 (3): e57639. doi:10.1371/journal.pone.0057639. PMC 3594230. PMID 23536766.
- ↑ Rotimi CN, Jorde LB (2010). "Ancestry and disease in the age of genomic medicine". N Engl J Med. 363 (16): 1551–8. doi:10.1056/NEJMra0911564. PMID 20942671.
- ↑ Bloche MG (2004). "Race-based therapeutics". N Engl J Med. 351 (20): 2035–7. doi:10.1056/NEJMp048271. PMID 15533852.
- ↑ Frank R (March 30 – April 1, 2006). "Back with a Vengeance: the Reemergence of a Biological Conceptualization of Race in Research on Race/Ethnic Disparities in Health". Annual Meeting of the Population Association of America. Los Angeles, California. Retrieved 2008-11-20.
- ↑ Crawley L (2007). "The paradox of race in the Bidil debate". J Natl Med Assoc. 99 (7): 821–2. PMC 2574363. PMID 17668653.
- ↑ Kalow, Werner (2005). Pharmacogenomics. New York: Taylor & Francis. pp. 552–3. ISBN 1-57444-878-1.
- ↑ Thorpe DS (2001). "Combinatorial chemistry: starting the second decade.". Pharmacogenomics J. 1 (4): 229–32. doi:10.1038/sj.tpj.6500045. PMID 11908762.
- ↑ Barh, Debmalya; Dhawan, Dipali; Ganguly, Nirmal Kumar (2013). Omics for Personalized Medicine. India: Springer Media. doi:10.1007/978-81-322-1184-6. ISBN 978-81-322-1183-9.
- ↑ Stram, Daniel (2014). Design, Analysis, and Interpretation of Genome-Wide Association Scans. Los Angeles: Springer Science and Business Media. doi:10.1007/978-1-4614-9443-0_8. ISBN 978-1-4614-9442-3.
Further reading
- Katsnelson A (August 2005). "A Drug to Call One's Own: Will medicine finally get personal?". Scientific American.
- Karczewski KJ, Daneshjou R, Altman RB (2012). "Chapter 7: Pharmacogenomics". PLoS Comput Biol. 8 (12): e1002817. doi:10.1371/journal.pcbi.1002817. PMC 3531317. PMID 23300409.
External links
- "Pharmacogenomics Factsheet". National Center for Biotechnology Information (NCBI), U.S. National Library of Medicine. Retrieved 2011-07-11.
a quick introduction to customised drugs
- "Pharmacogenomics Education Initiatives". U.S. Food and Drug Administration. 2010-09-24. Retrieved 2011-07-11.
- "Personalized Medicine (Pharmacogenetics)". University of Utah's Genetic Science Learning Center. Retrieved 2011-07-11.
- "Center for Pharmacogenomics and Individualized Therapy". University of North Carolina at Chapel Hill Center for Pharmacogenomics and Individualized Therapy. Retrieved 2014-06-25.
Journals:
- "Future Medicine - Pharmacogenomics". Journal. Future Medicine Ltd. ISSN 1462-2416.
- "Pharmacogenetics and Genomics". Journal (previously Pharmacogenetics). Lippincott Williams & Wilkins. ISSN 1744-6872. Retrieved 2011-07-11.
- "The Pharmacogenomics Journal". Nature Publishing Group. ISSN 1470-269X. Retrieved 2011-07-11.
- "Pharmacogenomics: Subjects : Omics Gateway". Nature Publishing Group. Retrieved 2011-07-11.