Periodic table (crystal structure)
For elements that are solid at standard temperature and pressure the table gives the crystalline structure of the most thermodynamically stable form(s) in those conditions. In all other cases the structure given is for the element at its melting point. Data is presented only for the first 112 elements (hydrogen through copernicium; it is not available for any further ones), and predictions are given for elements that have never been produced in bulk (astatine, francium, and elements 100–112).
Table
| Crystal structure of elements in the periodic table | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 H HEX |
2 He HCP | |||||||||||||||||
| 3 Li BCC |
4 Be HCP |
5 B RHO |
6 C HEX |
7 N HEX |
8 O SC |
9 F SC |
10 Ne FCC | |||||||||||
| 11 Na BCC |
12 Mg HCP |
13 Al FCC |
14 Si DC |
15 P ORTH |
16 S ORTH |
17 Cl ORTH |
18 Ar FCC | |||||||||||
| 19 K BCC |
20 Ca FCC |
21 Sc HCP |
22 Ti HCP |
23 V BCC |
24 Cr BCC |
25 Mn BCC |
26 Fe BCC |
27 Co HCP |
28 Ni FCC |
29 Cu FCC |
30 Zn HCP |
31 Ga ORTH |
32 Ge DC |
33 As RHO |
34 Se HEX |
35 Br ORTH |
36 Kr FCC | |
| 37 Rb BCC |
38 Sr FCC |
39 Y HCP |
40 Zr HCP |
41 Nb BCC |
42 Mo BCC |
43 Tc HCP |
44 Ru HCP |
45 Rh FCC |
46 Pd FCC |
47 Ag FCC |
48 Cd HCP |
49 In TETR |
50 Sn TETR |
51 Sb RHO |
52 Te HEX |
53 I ORTH |
54 Xe FCC | |
| 55 Cs BCC |
56 Ba BCC |
|
72 Hf HCP |
73 Ta BCC/TETR |
74 W BCC |
75 Re HCP |
76 Os HCP |
77 Ir FCC |
78 Pt FCC |
79 Au FCC |
80 Hg RHO |
81 Tl HCP |
82 Pb FCC |
83 Bi RHO |
84 Po SC/RHO |
85 At [FCC] |
86 Rn FCC | |
| 87 Fr [BCC] |
88 Ra BCC |
|
104 Rf [HCP] |
105 Db [BCC] |
106 Sg [BCC] |
107 Bh [HCP] |
108 Hs [HCP] |
109 Mt [FCC] |
110 Ds [BCC] |
111 Rg [BCC] |
112 Cn [HCP] |
113 Nh |
114 Fl |
115 Mc |
116 Lv |
117 Ts |
118 Og | |
| | ||||||||||||||||||
| |
57 La DHCP |
58 Ce DHCP/FCC |
59 Pr DHCP |
60 Nd DHCP |
61 Pm DHCP |
62 Sm RHO |
63 Eu BCC |
64 Gd HCP |
65 Tb HCP |
66 Dy HCP |
67 Ho HCP |
68 Er HCP |
69 Tm HCP |
70 Yb FCC |
71 Lu HCP | |||
| |
89 Ac FCC |
90 Th FCC |
91 Pa TETR |
92 U ORTH |
93 Np ORTH |
94 Pu MON |
95 Am DHCP |
96 Cm DHCP |
97 Bk DHCP |
98 Cf DHCP |
99 Es FCC |
100 Fm [FCC] |
101 Md [FCC] |
102 No [FCC] |
103 Lr [HCP] | |||
| Legend: |
|---|
| …/… mixed structure |
| […] predicted structure |
| unknown or uncertain |

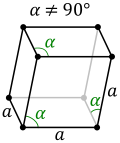
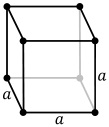
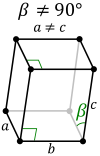
Unusual structures
| Element | crystal system | coordination number | notes |
|---|---|---|---|
| Mn | cubic | distorted bcc – unit cell contains Mn atoms in 4 different environments.[1] | |
| Zn | hexagonal | distorted from ideal hcp. 6 nearest neighbors in same plane- 6 in adjacent planes 14% farther away[1] | |
| Ga | orthorhombic | each Ga atom has one nearest neighbour at 244 pm, 2 at 270 pm, 2 at 273 pm, 2 at 279 pm.[1] | The structure is related to Iodine. |
| Cd | hexagonal | distorted from ideal hcp. 6 nearest neighbours in the same plane- 6 in adjacent planes 15% farther away[1] | |
| In | tetragonal | slightly distorted fcc structure[1] | |
| Sn | tetragonal | 4 neighbours at 302 pm; 2 at 318 pm; 4 at 377 pm; 8 at 441 pm [1] | white tin form (thermodynamical stable above 286.4 K) |
| Sb | rhombohedral | puckered sheet; each Sb atom has 3 neighbours in the same sheet at 290.8pm; 3 in adjacent sheet at 335.5 pm.[1] | grey metallic form. |
| Hg | rhombohedral | 6 nearest neighbours at 234 K and 1 atm (it is liquid at room temperature and thus has no crystal structure at ambient conditions!) | this structure can be considered to be a distorted hcp lattice with the nearest neighbours in the same plane being approx 16% farther away [1] |
| Bi | rhombohedral | puckered sheet; each Bi atom has 3 neighbours in the same sheet at 307.2 pm; 3 in adjacent sheet at 352.9 pm.[1] | Bi, Sb and grey As have the same space group in their crystal |
| Po | cubic | 6 nearest neighbours | simple cubic lattice. The atoms in the unit cell are at the corner of a cube. |
| Sm | trigonal | 12 nearest neighbours | complex hcp with 9 layer repeat, ABCBCACAB....[2] |
| Pa | tetragonal | body centred tetragonal unit cell, which can be considered to be a distorted bcc | |
| U | orthorhombic | strongly distorted hcp structure. Each atom has four near neighbours, 2 at 275.4 pm, 2 at 285.4 pm. The next four at distances 326.3 pm and four more at 334.2 pm.[3] | |
| Np | orthorhombic | highly distorted bcc structure. Lattice parameters: a=666.3 pm, b=472.3 pm, c=488.7 pm [4][5] | |
| Pu | monoclinic | slightly distorted hexagonal structure. 16 atoms per unit cell. Lattice parameters: a= 618.3 pm, b=482.2 pm, c=1096.3 pm, β= 101.79 ° [6][7] |
Usual crystal structures
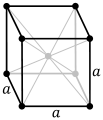
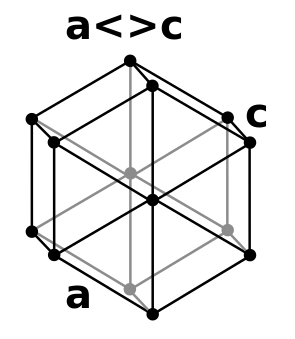
Close packed metal structures
Many metals adopt close packed structures i.e. hexagonal close packed and face centred cubic structures (cubic close packed). A simple model for both of these is to assume that the metal atoms are spherical and are packed together in the most efficient way (close packing or closest packing). In closest packing every atom has 12 equidistant nearest neighbours, and therefore a coordination number of 12. If the close packed structures are considered as being built of layers of spheres then the difference between hexagonal close packing and face centred cubic is how each layer is positioned relative to others. Whilst there are many ways that can be envisaged for a regular buildup of layers:
- hexagonal close packing has alternate layers positioned directly above/below each other, A,B,A,B, ......... (also termed P63/mmc, Pearson symbol hP2, strukturbericht A3) .
- face centered cubic has every third layer directly above/below each other,A,B,C,A,B,C,.......(also termed cubic close packing, Fm3m, Pearson symbol cF4, strukturbericht A1) .
- double hexagonal close packing has layers directly above/below each other, A,B,A,C,A,B,A,C,.... of period length 4 like an alternative mixture of fcc and hcp packing (also termed P63/mmc, Pearson Symbol hP4, strukturbericht A3' ).[8]
- α-Sm packing has a period of 9 layers A,B,A,B,C,B,C,A,C,.... (R3m, Pearson Symbol hR3, strukturbericht C19).[9]
Hexagonal close packed
In the ideal hcp structure the unit cell axial ratio is ~ 1.633, However, there are deviations from this in some metals where the unit cell is distorted in one direction but the structure still retains the hcp space group—remarkable all the elements have a ratio of lattice parameters c/a < 1.633 (best are Mg and Co and worst Be with c/a ~ 1.568). In others like Zn and Cd the deviations from the ideal change the symmetry of the structure and these have a lattice parameter ratio c/a > 1.85.

Face centered cubic (cubic close packed)
More content relating to number of planes within structure and implications for glide/slide e.g. ductility.
Double hexagonal close packed
Similar to the ideal hcp structure, the perfect dhcp structure should have a lattice parameter ratio of ~ 3.267. In the real dhcp structures of 5 lanthanides (including β-Ce) variates between 1.596 (Pm) and 1.6128 (Nd). For the 4 known actinides dhcp lattices the corresponding number vary between 1.620 (Bk) and 1.625 (Cf).[10]



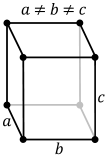
Body centred cubic
This is not a close packed structure. In this each metal atom is at the centre of a cube with 8 nearest neighbors, however the 6 atoms at the centres of the adjacent cubes are only approximately 15% further away so the coordination number can therefore be considered to be 14 when these are included. Note that if the body centered cubic unit cell is compressed along one 4 fold axis the structure becomes face centred cubic (cubic close packed).
See also
References
- 1 2 3 4 5 6 7 8 9 Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 0-08-037941-9.
- ↑ A.F Wells (1962) Structural Inorganic Chemistry 3d Edition Oxford University Press
- ↑ Harry L. Yakel, A REVIEW OF X-RAY DIFFRACTION STUDIES IN URANIUM ALLOYS. The Physical Metallurgy of Uranium Alloys Conference, Vail, Colorado, Feb. 1974
- ↑ Lemire,R.J. et al.,Chemical Thermodynamics of Neptunium and Plutonium, Elsevier, Amsterdam, 2001
- ↑ URL http://cst-www.nrl.navy.mil/lattice/struk/a_c.html
- ↑ Lemire,R.J. et al.,2001
- ↑ URL http://cst-www.nrl.navy.mil/lattice/struk/aPu.html
- ↑ URL http://cst-www.nrl.navy.mil/lattice/struk/a3p.html
- ↑ URL http://cst-www.nrl.navy.mil/lattice/struk/c19.html
- ↑ Nevill Gonalez Swacki & Teresa Swacka, Basic elements of Crystallography, Pan Standford Publishing Pte. Ltd., 2010
- General
- Actinides and the Environment, Edited by P.A. Sterne, A. Gonis and A.A. Borovoi, NATO ASI Series, Proc. of the NATO Advanced Study Institute on Actinides and the Environment, Maleme, Crete, Greece, July 1996, Kluver Academic Publishers,. pp. 59–61. ISBN 0-7923-4968-7.
- The Chemistry of the Actinide and Transactinide Elements, Edited by L.R. Morss, Norman M. Edelstein, Jean Fuger, 3rd. Edition, Springer 2007 ISBN 1402035551. ISBN 978-1402035555.
External links
- Strukturbericht Type A – structure reports for the pure elements
- Crystal Structures for the solid chemical elements at 1 bar
| Periodic table forms |
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sets of elements |
| ||||||||||||||||||||||||||||||
| Elements | |||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||
| See also | |||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||