Hyper-IgD syndrome
| Hyperimmunoglobulinemia D with recurrent fever | |
|---|---|
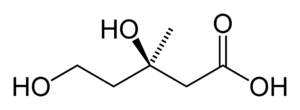 | |
| Mevalonic acid | |
| Classification and external resources | |
| OMIM | 260920 |
| DiseasesDB | 30161 |
Hyperimmunoglobulinemia D with recurrent fever (HIDS) is a periodic fever syndrome originally described in 1984 by the internist Jos van der Meer,[1] then at Leiden University Medical Centre. No more than 300 cases have been described worldwide.
Signs and symptoms
HIDS is one of a number of periodic fever syndromes. It is characterised by attacks of fever, arthralgia, skin lesions including cyclical mouth ulcers, and diarrhea. Laboratory features include an acute phase response (elevated CRP and ESR) and markedly elevated IgD (and often IgA), although cases with normal IgD have been described.[2]
It has mainly been described in the Netherlands and France, although the international registry includes a number of cases from other countries.[2]
The differential diagnosis includes fever of unknown origin, familial Mediterranean fever (FMF) and familial Hibernian fever (or TNFα reception associated periodic syndrome/TRAPS).[2]
Cause
Virtually all people with the syndrome have mutations in the gene for mevalonate kinase, which is part of the HMG-CoA reductase pathway, an important cellular metabolic pathway.[3][4] Indeed, similar fever attacks (but normal IgD) have been described in patients with mevalonic aciduria - an inborn error of metabolism now seen as a severe form of HIDS.[2]
Pathophysiology
It is not known how mevalonate kinase mutations cause the febrile episodes, although it is presumed that other products of the cholesterol biosynthesis pathyway, the prenylation chains (geranylgeraniol and farnesol) might play a role.[2]
Treatment
The recurring fevers are highly unpleasant for patients, but so far only the immunosuppressant drugs etanercept and anakinra[5] have been shown to be effective. Statin drugs might decrease the level of mevalonate and are presently being investigated. A recent single case report highlighted bisphosphonates as a potential therapeutic option.[6]
References
- ↑ van der Meer JW, Vossen JM, Radl J, et al. (May 1984). "Hyperimmunoglobulinaemia D and periodic fever: a new syndrome". Lancet. 1 (8386): 1087–90. doi:10.1016/S0140-6736(84)92505-4. PMID 6144826.
- 1 2 3 4 5 Drenth JP, van der Meer JW (December 2001). "Hereditary periodic fever". N. Engl. J. Med. 345 (24): 1748–57. doi:10.1056/NEJMra010200. PMID 11742050.
- ↑ Drenth JP, Cuisset L, Grateau G, et al. (June 1999). "Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group". Nat. Genet. 22 (2): 178–81. doi:10.1038/9696. PMID 10369262.
- ↑ Houten SM, Kuis W, Duran M, et al. (June 1999). "Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome". Nat. Genet. 22 (2): 175–7. doi:10.1038/9691. PMID 10369261.
- ↑ Rigante D, Ansuini V, Bertoni B, et al. (November 2006). "Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome". Rheumatol. Int. 27 (1): 97–100. doi:10.1007/s00296-006-0164-x. PMID 16871408.
- ↑ Cantarini, L; Vitale, A; Magnotti, F; Lucherini, O. M.; Caso, F; Frediani, B; Galeazzi, M; Rigante, D (2013). "Weekly oral alendronate in mevalonate kinase deficiency". Orphanet Journal of Rare Diseases. 8: 196. doi:10.1186/1750-1172-8-196. PMC 3880037
 . PMID 24360083.
. PMID 24360083.