Hereditary spherocytosis
| Hereditary spherocytosis | |
|---|---|
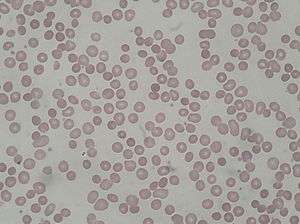 | |
| Peripheral blood smear from patient with hereditary spherocytosis | |
| Classification and external resources | |
| Specialty | hematology |
| ICD-10 | D58.0 |
| ICD-9-CM | 282.0 |
| OMIM | 182900 |
| DiseasesDB | 5827 |
| MedlinePlus | 000530 |
| eMedicine | med/2147 |
| MeSH | D013103 |
Hereditary spherocytosis (also known as Minkowski–Chauffard syndrome) is an autosomal dominant abnormality of erythrocytes. The disorder is caused by mutations in genes relating to membrane proteins that allow for the erythrocytes to change shape. The abnormal erythrocytes are sphere-shaped (spherocytosis) rather than the normal biconcave disk shaped. Dysfunctional membrane proteins interfere with the cell's ability to be flexible to travel from the arteries to the smaller capillaries. This difference in shape also makes the red blood cells more prone to rupture.[1] Cells with these dysfunctional proteins are taken for degradation at the spleen. This shortage of erythrocytes results in hemolytic anemia.
It was first described in 1871 and is the most common cause of inherited hemolysis in Europe and North America within the Caucasian population, with an incidence of 1 in 5000 births. The clinical severity of HS varies from symptom-free carrier to severe haemolysis because the disorder exhibits incomplete penetrance in its expression.
Symptoms include anemia, jaundice, splenomegaly, and fatigue. On a blood smear, Howell-Jolly bodies may be seen within red blood cells. Primary treatment for patients with symptomatic HS has been total splenectomy, which eliminates the hemolytic process, allowing normal hemoglobin, reticulocyte and bilirubin levels.
Signs and symptoms
As in non-hereditary spherocytosis, the spleen destroys the spherocytes. This process of red blood cells rupturing directly results in varying degrees of anemia (causing a pale appearance and fatigue), high levels of bilirubin in the blood (causing jaundice), and splenomegaly.
Acute cases can threaten to cause hypoxia through anemia and acute kernicterus through high blood levels of bilirubin, particularly in newborns. Most cases can be detected soon after birth. An adult with this disease should have their children tested, although the presence of the disease in children is usually noticed soon after birth. Occasionally, the disease will go unnoticed until the child is about 4 or 5 years of age. A person may also be a carrier of the disease and show no signs or symptoms of the disease. Other symptoms may include abdominal pain that could lead to the removal of the spleen and/or gallbladder.
Chronic symptoms include anemia, increased blood viscosity, and splenomegaly.[2] Furthermore, the detritus of the broken-down blood cells – unconjugated or indirect bilirubin – accumulates in the gallbladder, and can cause pigmented gallstones or "sludge" to develop. In chronic patients, an infection or other illness can cause an increase in the destruction of red blood cells, resulting in the appearance of acute symptoms, a hemolytic crisis. Spherocytosis patients who are heterozygous for a hemochromatosis gene may suffer from iron overload despite the hemochromatosis genes being recessive.[3][4]
Diagnosis
In a peripheral blood smear, the red blood cells will appear abnormally small and lack the central pale area that is present in normal red blood cells. These changes are also seen in non-hereditary spherocytosis, but they are typically more pronounced in hereditary spherocytosis. The number of immature red blood cells (reticulocyte count) will be elevated.[2] An increase in the mean corpuscular hemoglobin concentration is also consistent with hereditary spherocytosis.
Other protein deficiencies cause hereditary elliptocytosis, pyropoikilocytosis or stomatocytosis.
In longstanding cases and in patients who have taken iron supplementation or received numerous blood transfusions, iron overload may be a significant problem. This is a potential cause of heart muscle damage and liver disease. Measuring iron stores is therefore considered part of the diagnostic approach to hereditary spherocytosis.
An osmotic fragility test can aid in the diagnosis.[5] In this test, the spherocytes will rupture in liquid solutions less concentrated than the inside of the red blood cell. This is due to increased permeability of the spherocyte membrane to salt and water, which enters the concentrated inner environment of the RBC and leads to its rupture.[6] Although the osmotic fragility test is widely considered the gold standard for diagnosing hereditary spherocytosis, it misses as many as 25% of cases. Flow cytometric analysis of eosin-5′-maleimide-labeled intact red blood cells and the acidified glycerol lysis test are two additional options to aid diagnosis.[7]
Pathophysiology
Hereditary spherocytosis is an autosomal dominant or recessive trait,[8] most commonly (though not exclusively) found in Northern European and Japanese families, although an estimated 25% of cases are due to spontaneous mutations. A patient has a 50% chance of passing the mutation onto each of his/her offspring.
Hereditary spherocytosis is caused by a variety of molecular defects in the genes that code for the red blood cell proteins spectrin (alpha and beta), ankyrin,[9] band 3 protein, protein 4.2,[10] and other red blood cell membrane proteins:[11]
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| HS1 | 182900 | ANK1 | 8p11.2 |
| HS2 | 182870 | SPTB | 14q22-q23 |
| HS3 | 270970 | SPTA | 1q21 |
| HS4 | 109270 | SLC4A1 | 17q21-q22 |
| HS5 | 612690 | EPB42 | 15q15 |
These proteins are necessary to maintain the normal shape of a red blood cell, which is a biconcave disk. The integrating protein that is most commonly defective is ankyrin which is responsible for incorporation and binding of spectrin, thus in its dysfunction cytoskeletal instabilities ensue.
The primary defect in hereditary spherocytosis is a deficiency of membrane surface area. Decreased surface area may be produced by two different mechanisms: 1) Defects of spectrin, ankyrin (most commonly), or protein 4.2 lead to reduced density of the membrane skeleton, destabilizing the overlying lipid bilayer and releasing band 3-containing microvesicles. 2) Defects of band 3 lead to band 3 deficiency and loss of its lipid-stabilizing effect. This results in the loss of band 3-free microvesicles. Both pathways result in membrane loss, decreased surface area, and formation of spherocytes with decreased deformability.
As the spleen normally targets abnormally shaped red cells (which are typically older), it also destroys spherocytes. In the spleen, the passage from the cords of Billroth into the sinusoids may be seen as a bottleneck, where red blood cells need to be flexible in order to pass through. In hereditary spherocytosis, red blood cells fail to pass through and get phagocytosed, causing extravascular hemolysis.[12]
Complications
- Hemolytic crisis, with more pronounced jaundice due to accelerated hemolysis (may be precipitated by infection).
- Aplastic crisis with dramatic fall in hemoglobin level and (reticulocyte count)-decompensation, usually due to maturation arrest and often associated with megaloblastic changes; may be precipitated by infection, such as influenza, notably with parvovirus B19.[13][14][15]
- Folate deficiency caused by increased bone marrow requirement.
- Pigmented gallstones occur in approximately half of untreated patients. Increased hemolysis of red blood cells leads to increased bilirubin levels, because bilirubin is a breakdown product of heme. The high levels of bilirubin must be excreted into the bile by the liver, which may cause the formation of a pigmented gallstone, which is composed of calcium bilirubinate. Since these stones contain high levels of calcium carbonates and phosphate, they are radiopaque and are visible on x-ray.
- Leg ulcer.
- Abnormally low hemoglobin A1C levels.[16] Hemoglobin A1C (glycated hemoglobin) is a test for determining the average blood glucose levels over an extended period of time, and is often used to evaluate glucose control in diabetics. The hemoglobin A1C levels are abnormally low because the life span of the red blood cells is decreased, providing less time for the non-enzymatic glycosylation of hemoglobin. Thus, even with high overall blood sugar, the A1C will be lower than expected.
Treatment
At this point, there exists no cure for the genetic defect that causes hereditary spherocytosis.[11] Current management focuses on interventions that limit the severity of the disease. Treatment options include:
- Splenectomy: As in non-hereditary spherocytosis, acute symptoms of anemia and hyperbilirubinemia indicate treatment with blood transfusions or exchanges and chronic symptoms of anemia and splenomegaly indicate dietary supplementation of folic acid and splenectomy,[17] the surgical removal of the spleen. Splenectomy is indicated for moderate to severe cases, but not mild cases.[2] To decrease the risk of sepsis, post-splenectomy spherocytosis patients require immunization against the pneumococcus bacterium, influenza virus, and prophylactic antibiotic treatment. However, the use of prophylactic antibiotics, such as penicillin, remains controversial.[11]
- Partial splenectomy: Since the spleen is important for protecting against encapsulated organisms, sepsis caused by encapsulated organisms is a possible complication of splenectomy.[2] The option of partial splenectomy may be considered in the interest of preserving immune function. Research on outcomes is currently limited,[2] but favorable.[18]
- Cholecystectomy may be necessary.[11]
Epidemiology
Hereditary spherocytosis is the most common disorder of the red cell membrane and affects 1 in 2,000 people of Northern European ancestry. According to Harrison's Principles of Internal Medicine, the frequency is at least 1 in 5,000.[11]
Research
Experimental gene therapy exists to treat hereditary spherocytosis in lab mice; however, this treatment has not yet been tried on humans due to all of the risks involved in human gene therapy.
See also
References
- ↑ Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. p. 625. ISBN 0-7216-0187-1.
- 1 2 3 4 5 Bolton-Maggs, P. H. B.; Stevens, R. F.; Dodd, N. J.; Lamont, G.; Tittensor, P.; King, M. -J.; General Haematology Task Force of the British Committee for Standards in Haematology (2004). "Guidelines for the diagnosis and management of hereditary spherocytosis". British Journal of Haematology. 126 (4): 455–474. doi:10.1111/j.1365-2141.2004.05052.x. PMID 15287938.
- ↑ J L Rasmussen; D A Odelson; F L Macrina (1987-08-01). "Complete nucleotide sequence of insertion element IS4351 from Bacteroides fragilis. - UKPMC Article - UK PubMed Central". UKPMC Article. Retrieved 2012-07-03.
- ↑ Paula Bolton-Maggs (September 2011). "Guidelines for the Diagnosis and Management of Hereditary Spherocytosis" (PDF). The British Committee for Standards in Haematology. Retrieved 2 July 2012.
- ↑ Won DI, Suh JS (March 2009). "Flow cytometric detection of erythrocyte osmotic fragility". Cytometry Part B. 76 (2): 135–41. doi:10.1002/cyto.b.20448. PMID 18727072.
- ↑ Goljan. Rapid Review Pathology. 2010. Page 213.
- ↑ Bianchi, P.; Fermo, E.; Vercellati, C.; Marcello, A. P.; Porretti, L.; Cortelezzi, A.; Barcellini, W.; Zanella, A. (2011). "Diagnostic power of laboratory tests for hereditary spherocytosis: A comparison study in 150 patients grouped according to molecular and clinical characteristics". Haematologica. 97 (4): 516–523. doi:10.3324/haematol.2011.052845. PMC 3347664
 . PMID 22058213.
. PMID 22058213. - ↑ Eber S, Lux SE (April 2004). "Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer". Semin. Hematol. 41 (2): 118–41. doi:10.1053/j.seminhematol.2004.01.002. PMID 15071790.
- ↑ Gallagher PG, Forget BG (December 1998). "Hematologically important mutations: spectrin and ankyrin variants in hereditary spherocytosis". Blood Cells Mol. Dis. 24 (4): 539–43. doi:10.1006/bcmd.1998.0217. PMID 9887280.
- ↑ Perrotta S, Gallagher PG, Mohandas N (October 2008). "Hereditary spherocytosis". Lancet. 372 (9647): 1411–26. doi:10.1016/S0140-6736(08)61588-3. PMID 18940465.
- 1 2 3 4 5 Anthony S. Fauci; Eugene Braunwald; Dennis L. Kasper; Stephen L. Hauser; Dan L. Longo; J. Larry Jameson; Joseph Loscalzo (2008). Harrison's principles of internal medicine (17th ed.). New York: McGraw-Hill Medical. pp. Chapter 106. ISBN 978-0071466332.
- ↑ Chapter 12, page 425 in: Mitchell, Richard Sheppard; Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson. Robbins Basic Pathology. Philadelphia: Saunders. ISBN 1-4160-2973-7. 8th edition.
- ↑ Fjaerli, H. O.; Vogt, H.; Bruu, A. L. (1991). "Human parvovirus B19 as the cause of aplastic crisis in hereditary spherocytosis". Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke. 111 (22): 2735–2737. PMID 1658972.
- ↑ Beland, S. S.; Daniel, G. K.; Menard, J. C.; Miller, N. M. (1997). "Aplastic crisis associated with parvovirus B19 in an adult with hereditary spherocytosis". The Journal of the Arkansas Medical Society. 94 (4): 163–164. PMID 9308316.
- ↑ Servey, J. T.; Reamy, B. V.; Hodge, J. (2007). "Clinical presentations of parvovirus B19 infection". American family physician. 75 (3): 373–376. PMID 17304869.
- ↑ Kutter, D; Thoma, J (2006). "Hereditary spherocytosis and other hemolytic anomalies distort diabetic control by glycated hemoglobin.". Clinical laboratory. 52 (9-10): 477–81. PMID 17078474.
- ↑ Bolton-Maggs PH, Stevens RF, Dodd NJ, Lamont G, Tittensor P, King MJ (August 2004). "Guidelines for the diagnosis and management of hereditary spherocytosis". Br. J. Haematol. 126 (4): 455–74. doi:10.1111/j.1365-2141.2004.05052.x. PMID 15287938.
- ↑ Buesing, K. L.; Tracy, E. T.; Kiernan, C.; Pastor, A. C.; Cassidy, L. D.; Scott, J. P.; Ware, R. E.; Davidoff, A. M.; Rescorla, F. J.; Langer, J. C.; Rice, H. E.; Oldham, K. T. (2011). "Partial splenectomy for hereditary spherocytosis: A multi-institutional review". Journal of Pediatric Surgery. 46 (1): 178–183. doi:10.1016/j.jpedsurg.2010.09.090. PMID 21238662.
External links
- An online HS resource from The University of Texas Southwestern Medical Center
- A short article from WebMD
- A picture of spherocytes from Medline