Phase rule
Gibbs' phase rule[1][2] was proposed by Josiah Willard Gibbs in his landmark paper titled On the Equilibrium of Heterogeneous Substances, published from 1875 to 1878. The rule applies to non-reactive multi-component heterogeneous systems in thermodynamic equilibrium and is given by the equality

where F is the number of degrees of freedom, C is the number of components and P is the number of phases in thermodynamic equilibrium with each other.
The number of degrees of freedom is the number of independent intensive variables, i.e. the largest number of thermodynamic parameters such as temperature or pressure that can be varied simultaneously and arbitrarily without affecting one another. An example of one-component system is a system involving one pure chemical, while two-component systems, such as mixtures of water and ethanol, have two chemically independent components, and so on. Typical phases are solids, liquids and gases.
Foundations
- A phase is a form of matter that is homogeneous in chemical composition and physical state. Typical phases are solid, liquid and gas. Two immiscible liquids (or liquid mixtures with different compositions) separated by a distinct boundary are counted as two different phases, as are two immiscible solids.
- The number of components (C) is the number of chemically independent constituents of the system, i.e. the minimum number of independent species necessary to define the composition of all phases of the system.[2] For examples see component (thermodynamics).
- The number of degrees of freedom (F) in this context is the number of intensive variables which are independent of each other.
The basis for the rule (Atkins and de Paula,[2] justification 6.1) is that equilibrium between phases places a constraint on the intensive variables. More rigorously, since the phases are in thermodynamic equilibrium with each other, the chemical potentials of the phases must be equal. The number of equality relationships determines the number of degrees of freedom. For example, if the chemical potentials of a liquid and of its vapour depend on temperature (T) and pressure (p), the equality of chemical potentials will mean that each of those variables will be dependent on the other. Mathematically, the equation μliq(T, p) = μvap(T, p), where μ = chemical potential, defines temperature as a function of pressure or vice versa. (Caution: do not confuse p = pressure with P = number of phases.)
To be more specific, the composition of each phase is determined by C − 1 intensive variables (such as mole fractions) in each phase. The total number of variables is (C − 1)P + 2, where the extra two are temperature T and pressure p. The number of constraints is C(P − 1), since the chemical potential of each component must be equal in all phases. Subtract the number of constraints from the number of variables to obtain the number of degrees of freedom as F = (C − 1)P + 2 − C(P − 1) = C − P + 2.
The rule is valid provided the equilibrium between phases is not influenced by gravitational, electrical or magnetic forces, or by surface area, and only by temperature, pressure, and concentration.
Consequences and examples
Pure substances (one component)
For pure substances C = 1 so that F = 3 − P. In a single phase (P = 1) condition of a pure component system, two variables (F = 2), such as temperature and pressure, can be chosen independently to be any pair of values consistent with the phase. However, if the temperature and pressure combination ranges to a point where the pure component undergoes a separation into two phases (P = 2), F decreases from 2 to 1. When the system enters the two-phase region, it becomes no longer possible to independently control temperature and pressure.

In the phase diagram to the right, the boundary curve between the liquid and gas regions maps the constraint between temperature and pressure when the single-component system has separated into liquid and gas phases at equilibrium. If the pressure is increased by compression, some of the gas condenses and the temperature goes up. If the temperature is decreased by cooling, some of the gas condenses, decreasing the pressure. Throughout both processes, the temperature and pressure stay in the relationship shown by this boundary curve unless one phase is entirely consumed by evaporation or condensation, or unless the critical point is reached. As long as there are two phases, there is only one degree of freedom, which corresponds to the position along the phase boundary curve.
The critical point is the black dot at the end of the liquid–gas boundary. As this point is approached, the liquid and gas phases become progressively more similar until, at the critical point, there is no longer a separation into two phases. Above the critical point and away from the phase boundary curve, F = 2 and the temperature and pressure can be controlled independently. Hence there is only one phase, and it has the physical properties of a dense gas, but is also referred to as a supercritical fluid.
Of the other two-boundary curves, one is the solid–liquid boundary or melting point curve which indicates the conditions for equilibrium between these two phases, and the other at lower temperature and pressure is the solid–gas boundary.
Even for a pure substance, it is possible that three phases, such as solid, liquid and vapour, can exist together in equilibrium (P = 3). If there is only one component, there are no degrees of freedom (F = 0) when there are three phases. Therefore, in a single-component system, this three-phase mixture can only exist at a single temperature and pressure, which is known as a triple point. Here there are two equations μsol(T, p) = μliq(T, p) = μvap(T, p), which are sufficient to determine the two variables T and p. In the diagram for CO2 the triple point is the point at which the solid, liquid and gas phases come together, at 5.2 bar and 217 K. It is also possible for other sets of phases to form a triple point, for example in the water system there is a triple point where ice I, ice III and liquid can coexist.
If four phases of a pure substance were in equilibrium (P = 4), the phase rule would give F = −1, which is meaningless, since there cannot be −1 independent variables. This explains the fact that four phases of a pure substance (such as ice I, ice III, liquid water and water vapour) are not found in equilibrium at any temperature and pressure. In terms of chemical potentials there are now three equations, which cannot in general be satisfied by any values of the two variables T and p, although in principle they might be solved in a special case where one equation is mathematically dependent on the other two. In practice, however, the coexistence of more phases than allowed by the phase rule normally means that the phases are not all in true equilibrium.
Two-component systems
For binary mixtures of two chemically independent components, C = 2 so that F = 4 − P. In addition to temperature and pressure, the other degree of freedom is the composition of each phase, often expressed as mole fraction or mass fraction of one component.
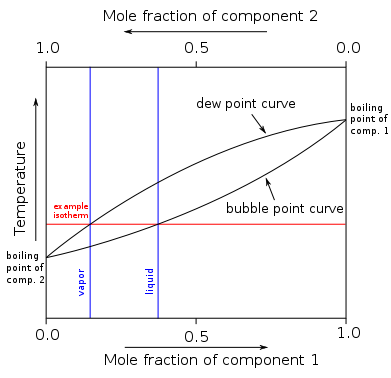
As an example, consider the system of two completely miscible liquids such as toluene and benzene, in equilibrium with their vapours. This system may be described by a boiling-point diagram which shows the composition (mole fraction) of the two phases in equilibrium as functions of temperature (at a fixed pressure).
Four thermodynamic variables which may describe the system include temperature (T), pressure (p), mole fraction of component 1 (toluene) in the liquid phase (x1L), and mole fraction of component 1 in the vapour phase (x1V). However since two phases are in equilibrium, only two of these variables can be independent (F = 2). This is because the four variables are constrained by two relations: the equality of the chemical potentials of liquid toluene and toluene vapour, and the corresponding equality for benzene.
For given T and p, there will be two phases at equilibrium when the overall composition of the system (system point) lies in between the two curves. A horizontal line (isotherm or tie line) can be drawn through any such system point, and intersects the curve for each phase at its equilibrium composition. The quantity of each phase is given by the lever rule (expressed in the variable corresponding to the x-axis, here mole fraction).
For the analysis of fractional distillation, the two independent variables are instead considered to be liquid-phase composition (x1L) and pressure. In that case the phase rule implies that the equilibrium temperature (boiling point) and vapour-phase composition are determined.
Liquid–vapour phase diagrams for other systems may have azeotropes (maxima or minima) in the composition curves, but the application of the phase rule is unchanged. The only difference is that the compositions of the two phases are equal exactly at the azeotropic composition.
Phase rule at constant pressure
For applications in materials science dealing with phase changes between different solid structures, pressure is often imagined to be constant (for example at one atmosphere), and is ignored as a degree of freedom, so the rule becomes
- F = C − P + 1.
This is sometimes misleadingly called the "condensed phase rule", but it is not applicable to condensed systems which are subject to high pressures (for example, in geology), since the effects of these pressures can be important.
See also
References
Further reading
- Mogk, David: Teaching Phase Equilibria. Gibbs' Phase Rule: Where it all Begins (The phase rule in geology)
- Predel, Bruno; Hoch, Michael J. R.; Pool, Monte. Phase Diagrams and Heterogeneous Equilibria : A Practical Introduction. Springer. ISBN 3-540-14011-5.
- White, Mary Anne. Properties of Materials. Oxford University Press (1999). ISBN 0-19-511331-4. Chapter 9. Thermodynamics Aspects of Stability