Friedreich's ataxia
| Friedreich's ataxia | |
|---|---|
 | |
| Frataxin | |
| Classification and external resources | |
| Specialty | neurology |
| ICD-10 | G11.1 |
| ICD-9-CM | 334.0 |
| OMIM | 229300 |
| DiseasesDB | 4980 |
| MedlinePlus | 001411 |
| eMedicine | article/1150420 |
| Patient UK | Friedreich's ataxia |
| MeSH | D005621 |
| GeneReviews | |
Friedreich's ataxia is an autosomal recessive inherited disease that causes progressive damage to the nervous system. It manifests in initial symptoms of poor coordination such as gait disturbance; it can also lead to scoliosis, heart disease and diabetes, but does not affect cognitive function. The disease progresses until a wheelchair is required for mobility. Its incidence in the general population is roughly 1 in 50,000.
The particular genetic mutation (expansion of an intronic GAA triplet repeat in the FXN gene) leads to reduced expression of the mitochondrial protein frataxin. Over time this deficiency causes the aforementioned damage, as well as frequent fatigue due to effects on cellular metabolism.
The ataxia of Friedreich's ataxia results from the degeneration of nervous tissue in the spinal cord, in particular sensory neurons essential (through connections with the cerebellum) for directing muscle movement of the arms and legs. The spinal cord becomes thinner and nerve cells lose some of their myelin sheath (the insulating covering on some nerve cells that helps conduct nerve impulses).
The condition is named after the German physician Nikolaus Friedreich, who first described it in the 1860s.[1]
Signs and symptoms
Symptoms typically begin sometime between the ages of 5 to 15 years, but in Late Onset FA may occur in the 20s or 30s. Symptoms include any combination, but not necessarily all, of the following:
- Muscle weakness in the arms and legs
- Loss of coordination
- Vision impairment
- Hearing impairment
- Slurred speech
- Curvature of the spine (scoliosis)
- High plantar arches (pes cavus deformity of the foot)
- Diabetes (about 20% of people with Friedreich's ataxia develop carbohydrate intolerance and 10% develop diabetes mellitus)[2]
- Heart disorders (e.g., atrial fibrillation, and resultant tachycardia (fast heart rate) and hypertrophic cardiomyopathy)
It presents before 22 years of age with progressive staggering or stumbling gait and frequent falling. Lower extremities are more severely involved. The symptoms are slow and progressive. Long-term observation shows that many patients reach a plateau in symptoms in the patient's early adulthood. On average, after 10–15 years with the disease, patients are usually wheelchair bound and require assistance with all activities of daily living.[3]
The following physical signs may be detected on physical examination:
- Cerebellar: Nystagmus, fast saccadic eye movements, truncal ataxia, dysarthria, dysmetria.
- Lower motor neuron lesion: absent deep tendon reflexes.
- Pyramidal: extensor plantar responses, and distal weakness are commonly found.
- Dorsal column: Loss of vibratory and proprioceptive sensation occurs.
- Cardiac involvement occurs in 91% of patients, including cardiomegaly (up to dilated cardiomyopathy), symmetrical hypertrophy, heart murmurs, and conduction defects. Median age of death is 35 years, while females have better prognosis with a 20-year survival of 100% as compared to 63% in men.
20% of cases are found in association with diabetes mellitus.[2]
Genetics
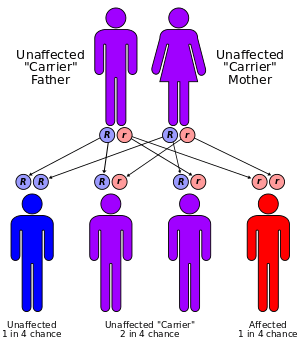
Friedreich's ataxia is an autosomal recessive disorder that occurs when the FXN gene contains amplified intronic GAA repeats (an example of Trinucleotide repeat expansion). The FXN gene encodes the protein frataxin.[4] GAA repeat expansion causes frataxin levels to be reduced. Frataxin is an iron-binding protein responsible for forming iron–sulphur clusters. One result of frataxin deficiency is mitochondrial iron overload which can cause damage to many proteins.[4] The exact role of frataxin in normal physiology remains unclear.[5] The gene is located on chromosome 9.
The mutant gene contains expanded GAA triplet repeats in the first intron;[6] in a few pedigrees, point mutations have been detected. Because the defect is located in an intron (which is removed from the mRNA transcript between transcription and translation), this mutation does not result in the production of abnormal frataxin proteins. Instead, the mutation causes gene silencing (i.e., the mutation decreases the transcription of the gene) through induction of a heterochromatin structure in a manner similar to position-effect variegation.[7]
Besides reducing expression of frataxin, long tracts of GAA repeats induce chromosome breaks in in vivo yeast studies.
Pathology
The primary site of pathology is spinal cord and peripheral nerves. Sclerosis and degeneration of dorsal root ganglion, spinocerebellar tracts, lateral corticospinal tracts, and posterior columns.[8] The motor neurons of the spinal cord are spared. In peripheral nerves there is a loss of large myelinated fibres.
Progressive destruction of dorsal root ganglia accounts for thinning of dorsal roots, degeneration of dorsal columns, transsynaptic atrophy of nerve cells in Clarke's column and dorsal spinocerebellar fibers, atrophy of gracile and cuneate nuclei and neuropathy of sensory nerves. The lesion of the dentate nucleus consists of progressive and selective atrophy of large glutamatergic neurons and grumose degeneration of corticonuclear synaptic terminals that contain gamma-aminobutyric acid (GABA). Small GABA-ergic neurons and their projection fibers in the dentato-olivary tract survive. Atrophy of Betz cells and corticospinal tracts constitute a second lesion.
Pathogenesis
Low frataxin levels lead to insufficient biosynthesis of iron-sulfur clusters that are required for mitochondrial electron transport and assembly of functional aconitase and iron dysmetabolism of the entire cell. In normal individuals, the FXN gene encodes frataxin, a mitochondrial matrix protein. This globular protein consists of two α helices and seven β strands and is highly conserved, occurring in all eukaryotes and some prokaryotes.[9] Frataxin has a variety of known functions. Frataxin assists iron-sulfur protein synthesis in the electron transport chain to ultimately generate adenosine triphosphate (ATP), the energy currency necessary to carry out metabolic functions in cells. Frataxin also regulates iron transfer in the mitochondria in order provide a proper amount of reactive oxygen species (ROS) to maintain normal processes.[10] Without frataxin, the energy in the mitochondria fails, and excess iron causes extra ROS to be created, leading to further cell damage.[9][10]
Treatment
A person suffering from Friedreich's Ataxia may require some surgical interventions (mainly for the spine and heart). Often, titanium screws and rods are inserted in the spine to help prevent or slow the progression of scoliosis. As progression of ataxia occurs, assistive devices such as a cane, walker, or wheelchair are required for mobility and independence. Other assistive technology, such as a standing frame, can help reduce the secondary complications of prolonged use of a wheelchair. The goal of surgery is to keep the patient ambulatory as long as possible.
In many cases, patients experience significant heart conditions as well. These conditions are much more treatable, and are often countered with ACE inhibitors such as enalapril or lisinopril and other heart medications such as digoxin.
Persons with Friedreich’s ataxia may also benefit from a conservative treatment approach for the management of symptoms. Health professionals educated in neurological conditions, such as physical therapists and occupational therapists, can prescribe an exercise program tailored to maximize function and independence. To address the ataxic gait pattern and loss of proprioception typically seen in persons with Friedreich’s ataxia, physical therapists can use visual cueing during gait training to help facilitate a more efficient gait pattern.[11] The prescription of an assistive device along with gait training can also prolong independent ambulation.[11]
Low intensity strengthening exercises should also be incorporated to maintain functional use of the upper and lower extremities.[12] Fatigability should be monitored closely. Stabilization exercises of the trunk and low back can help with postural control and the management of scoliosis.[11] This is especially indicative if the person is non-ambulatory and requires the use of a wheelchair. Balance and coordination training using visual feedback can also be incorporated into activities of daily living.[11] Exercises should reflect functional tasks such as cooking, transfers and self-care. Along with gait training, balance and coordination training should be developed to help minimize the risk of falls.[11]
Stretching exercises can be prescribed to help relieve tight musculature due to scoliosis and pes cavus deformities.[12]
Idebenone
Idebenone, an antioxidant, was recently removed from the Canadian market in 2013 due to lack of effectiveness.[13]
A Cochrane review on antioxidants and other pharmacological treatment of patients with Friedreich Ataxia, first published in 2012, is due to be updated before the end of 2015.[14]
Nicotinamide
Nicotinamide administration on patients was associated with a sustained improvement in frataxin concentrations towards those seen in asymptomatic carriers during 8 weeks of daily dosing. The daily oral administration of 3.8 g nicotinamide resulted in a 1.5-times increase, whereas 7.5 g resulted in a doubling of frataxin protein concentration.[15]
Speech therapy
Patients also often undertake speech therapy since dysarthria (a motor speech disorder) occurs in almost 100% of Friedreich's ataxia patients. However, the dysarthria is not always ataxic and the dysarthria can be mixed. The speech intelligibility in speakers with dysarthria and Friedreich's Ataxia can be mild to severely reduced. Speech therapy seeks to improve speech outcomes and/or compensate for communication deficits.[16] Dysphagia (difficulty swallowing) is also a common symptom of Friedreich's ataxia, and speech therapy can support patients to eat and drink in a safer way.[17]
Clinical research
RG2833, a histone deacetylase inhibitor developed by Repligen, was acquired by BioMarin Pharmaceutical in January 2014.[18] The first human trials with this compound began in 2012.[19]
Horizon Pharma's development plan of interferon gamma-1B for treatment of FA was given fast track designation by the Food and Drug Administration in 2015.[20]
Epidemiology
Friedreich's ataxia is the most prevalent inherited ataxia,[21] affecting about 1 in 50,000 people in the United States. Males and females are affected equally. The estimated carrier prevalence is 1:110.
A 1984 Canadian study was able to trace 40 cases of classical Friedreich's disease from 14 French-Canadian kindreds previously thought to be unrelated to one common ancestral couple arriving in New France in 1634: Jean Guyon and Mathurine Robin.[22]
History
Friedreich, working as a professor of pathology at the University of Heidelberg, reported five patients with the condition in a series of three papers in 1863.[23][24][25] Further observations appeared in a subsequent paper in 1876.[26]
References
- ↑ synd/1406 at Who Named It?
- 1 2 Thoren, Claes (June 1962). "Diabetes Mellitus in Friedreich's Ataxia". Acta Paediatrica. 51: 239–247. doi:10.1111/j.1651-2227.1962.tb08680.x. PMID 13921008.
- ↑ Pandolfo M (2009). "Friedreich ataxia: The clinical picture". Journal of Neurology, 256: (1 Suppl), 3-8, doi: 10.1007/s00415-009-1002-3
- 1 2 Klockgether T (August 2011). "Update on degenerative ataxias". Curr. Opin. Neurol. 24 (4): 339–45. doi:10.1097/WCO.0b013e32834875ba. PMID 21734495.
- ↑ Marmolino, D. (2011). "Friedreich's ataxia: Past, present and future". Brain Research Reviews. 67 (1–2): 311–330. doi:10.1016/j.brainresrev.2011.04.001. PMID 21550666.
- ↑ Montermini L, Andermann E, Labuda M, et al. (August 1997). "The Friedreich ataxia GAA triplet repeat: premutation and normal alleles". Hum. Mol. Genet. 6 (8): 1261–6. doi:10.1093/hmg/6.8.1261. PMID 9259271.
- ↑ Friedreich Ataxia at eMedicine
- ↑ Delatycki M, Williamson R, Forrest S (2000). "Friedreich ataxia: an overview". J Med Genet. 37 (1): 1–8 As. doi:10.1136/jmg.37.1.1. PMC 1734457
 . PMID 10633128.
. PMID 10633128. - 1 2 Pandolfo, M (October 2008). "Friedreich ataxia.". Archives of neurology. 65 (10): 1296–303. doi:10.1001/archneur.65.10.1296. PMID 18852343.
- 1 2 Sahdeo, S.; Scott, B. D.; McMackin, M. Z.; Jasoliya, M.; Brown, B.; Wulff, H.; Perlman, S. L.; Pook, M. A.; Cortopassi, G. A. (11 August 2014). "Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich's ataxia". Human Molecular Genetics. 23: 6848–62. doi:10.1093/hmg/ddu408. PMID 25113747.
- 1 2 3 4 5 Powers, Wendy (2007-01-01). "Holding Steady: How physical therapy can help patients with Friedreich's Ataxia". Advance. 18 (1): 26. Retrieved 2011-05-16.
- 1 2 "Facts About Friedreich's Ataxia (FA)". Muscular Dystrophy Association. 2011. Retrieved 2011-05-16.
- ↑ "CATENA (idebenone) - Voluntary withdrawal of CATENA from the Canadian market - For the Public - Recalls & alerts - Healthy Canadians Website". Healthycanadians.gc.ca. Retrieved 2015-07-04.
- ↑ Kearney, Mary; Orrell, Richard W; Fahey, Michael; Pandolfo, Massimo; Kearney, Mary (2012). "Antioxidants and other pharmacological treatments for Friedreich ataxia". The Cochrane Database of Systematic Reviews. 4: CD007791. doi:10.1002/14651858.CD007791.pub3. PMID 22513953.
- ↑ Libri, Vincenzo; Yandim, Cihangir; Athanasopoulos, Stavros; Loyse, Naomi; Natisvili, Theona; Law, Pui Pik; Chan, Ping Kei; Mohammad, Tariq; Mauri, Marta; Tam, Kin Tung; Leiper, James; Piper, Sophie; Ramesh, Aravind; Parkinson, Michael H; Huson, Les; Giunti, Paola; Festenstein, Richard (2014). "Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich's ataxia: an exploratory, open-label, dose-escalation study". The Lancet. 384 (9942): 504–13. doi:10.1016/S0140-6736(14)60382-2. PMID 24794816.
- ↑ Vogel, Adam P; Folker, Joanne; Poole, Matthew L; Vogel, Adam P (2014). "Treatment for speech disorder in Friedreich ataxia and other hereditary ataxia syndromes". The Cochrane Database of Systematic Reviews. 10: CD008953. doi:10.1002/14651858.CD008953.pub2. PMID 25348587.
- ↑ Vogel, Adam P.; Brown, Sophie E.; Folker, Joanne E.; Corben, Louise A.; Delatycki, Martin B. (2014-02-01). "Dysphagia and swallowing-related quality of life in Friedreich ataxia". Journal of Neurology. 261 (2): 392–399. doi:10.1007/s00415-013-7208-4. ISSN 1432-1459. PMID 24371004.
- ↑ "BioMarin Announces Agreement With Repligen for Pre-clinical Compounds (NASDAQ:BMRN)". Investors.bmrn.com. 2014-01-21. Retrieved 2015-07-04.
- ↑ "FARA Human Trials" (PDF). Friedreich's Ataxia Research Alliance. 2012. Retrieved 2012-03-15.
- ↑ Dulaney, Chelsey (2015-04-10). "Horizon Pharma's Friedreich's Ataxia Drug Gets Fast-Track Designation". The Wall Street Journal. Retrieved 2015-07-04.
- ↑ Lodi R, Tonon C, Calabrese V, Schapira AH (2006). "Friedreich's ataxia: from disease mechanisms to therapeutic interventions". Antioxid. Redox Signal. 8 (3–4): 438–43. doi:10.1089/ars.2006.8.438. PMID 16677089.
- ↑ Barbeau A, Sadibelouiz M, Roy M, Lemieux B, Bouchard JP, Geoffroy G (1984). "Origin of Friedreich's disease in Quebec". The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiques. 11 (4 Suppl): 506–9. PMID 6391645.
- ↑ Friedreich N (1863). "Ueber degenerative Atrophie der spinalen Hinterstränge" [About degenerative atrophy of the spinal posterior column]. Arch Pathol Anat Phys Klin Med (in German). 26 (3–4): 391–419. doi:10.1007/BF01930976.
- ↑ Friedreich N (1863). "Ueber degenerative Atrophie der spinalen Hinterstränge" [About degenerative atrophy of the spinal posterior column]. Arch Pathol Anat Phys Klin Med (in German). 26 (5–6): 433–459. doi:10.1007/BF01878006.
- ↑ Friedreich N (1863). "Ueber degenerative Atrophie der spinalen Hinterstränge" [About degenerative atrophy of the spinal posterior column]. Arch Pathol Anat Phys Klin Med (in German). 27 (1–2): 1–26. doi:10.1007/BF01938516.
- ↑ Friedreich N (1876). "Ueber Ataxie mit besonderer Berücksichtigung der hereditären Formen" [About ataxia with special reference to hereditary forms]. Arch Pathol Anat Phys Klin Med (in German). 68 (2): 145–245. doi:10.1007/BF01879049.
External links
- European Friedreich's Ataxia Consortium for Translational Studies
- friedreichs_ataxia at NINDS
- friedreich at NIH/UW GeneTests
- FARA The Friedreich's Ataxia Research Alliance at www.cureFA.org
- Asks the Experts - Responses: Friedreich's Ataxia at Muscular Dystrophy Association
- NCBI Genes and Disease: Friedreich's ataxia at National Center for Biotechnology Information
- Canadian Association for Familial Ataxias - Claude St-Jean Foundation
- British Columbia (BC) Ataxia Society