Discovery and development of memantine and related compounds
In 1972 a possible therapeutic importance of memantine, an adamantane derivative, was discovered for treating neurodegenerative disorders. Since 1989 memantine has been recognized to be an uncompetitive antagonist of the N-methyl-D-aspartate receptor (NMDA receptor), entering the channel of the receptor after it has been activated and thereby blocking the flow of ions.[1][2][3]
The NMDA receptor channels play an important role in synaptic plasticity and synapse formation underlying memory, learning and formation of neural networks during development in the central nervous system (CNS). Overactivation of the receptor, causing excessive influx of Ca2+ can lead to excitotoxicity which is implied to be involved in some neurodegenerative disorders. Blocking of NMDA receptors could therefore, in theory, be useful in treating such diseases.[3][4][5][6]
The main problem with the development of NMDA receptor antagonist for neuroprotection is that the physiological NMDA receptor activity is essential for normal neuronal function. To be clinically accepted the antagonists must block excessive activation without blocking the normal function.[7]
History
The discovery of NMDA receptors was followed by the synthesis and study of N-Methyl-D-aspartic acid (NMDA) in the 1960s by Jeff Watkins and colleagues. In the early 1980s, NMDA receptors were shown to be involved in several central synaptic pathways.[8][9] Receptor subunit selectivity was discovered in the early 1990s, which led to recognition of a new class of compounds that selectively inhibit the NR2B subunit. These findings led to vigorous campaign in the pharmaceutical industry.[10] From this it was considered that NMDA receptors were associated with a variety of neurological disorders such as epilepsy, Parkinson´s, Alzheimer´s, Huntington´s and other CNS disorders.[11]
A fortuitous finding was made in 1968 when a woman was taking amantadine as flu medicine and experienced remarkable remission of her Parkinson's symptoms. This finding, reported by Scawab et al., was the beginning of medicinal chemistry of adamantane derivatives in the context of diseases affecting the CNS.[12] Before this finding, memantine, another adamantane derivative, had been synthesized by Eli Lilly and Company in 1963. The purpose was to develop hypoglycemic drug, but it showed no such efficacy. It was not until 1972 that a possible therapeutic importance of memantine for treating neurodegenerative disorders was discovered. From 1989 memantine has been recognized to be an uncompetitive antagonist of the NMDA receptor.[2]
NMDA receptor

The NMDA receptor is a glutamate and ion channel protein receptor that is activated when glycine and glutamate bind to it.[13] The receptor is a heteromeric complex that interacts with multiple intracellular proteins by three different subunits: NR1, NR2 and NR3. NR1 has eight different subunits generated by alternative splicing from a single gene. There are four different NR2 subunits (A-D) and late in the 20 century NR3A and NR3B subunits have been reported. Six separate genes encode for NR2 and NR3.[14][15] All the subunits share a common membrane topology that is dominated by a large extracellular N-terminus, a membrane region comprising three transmembrane segments, a re-entrant pore loop, an extracellular loop between the transmembrane segments that are structurally not well known, and an intracellular C-terminus, which are different in size depending on the subunit and provide multiple sites of interaction with many intracellular proteins.[14][16] Figure 1 shows a basic structure of NR1/NR2 subunits that forms the binding site for memantine, Mg2+, MK-801, ketamine and amantadine.
Mg2+ block the NMDA receptor channels in voltage dependent manner but they are highly permeable to Ca2+. Activation of the receptor depends on glutamate binding, D-serine or glycine binding at its NR1-linked binding site and AMPA receptor-mediated depolarization of the postsynaptic membrane, which relieves the voltage-dependent channel block by Mg2+. Activation and opening of the receptors channel thus allows the flow of K+, Na+ and Ca2+ ions, and the influx of Ca2+ triggers intracellular signaling pathways.[1][17] Allosteric receptor binding sites for zinc, proteins and the polyamines spermidine and spermine are also modulators for the NMDA receptor channels.[18]
The NR2B subunit has been involved in modulating activity such as learning, memory, processing and feeding behaviors, as well as being implicated in number of human derangement. The basic structure and functions associated with the NMDA receptor can be attributed to the NR2B subunit. For example, the glutamate binding site and the control of the Mg2+ block are formed by the NR2B subunit. The high affinity sites for glycine antagonist are also exclusively displayed by the NR1/NR2B receptor.[15]
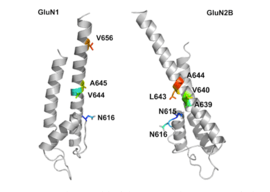
NR1/NR2B transmembrane segments are considered to be the part of the receptor that forms the binding pockets for uncompetitive NMDA receptor antagonists, but the transmembrane segments structures are not fully known as stated above. It is claimed that three binding sites within the receptor, A644 on the NR2B subunit and A645 and N616 on the NR1 subunit, are important for binding of memantine and related compounds as seen in figure 2.[16]
Mechanism of action
Overactivation of NMDA receptors, relieving the Mg2+ block and causing excessive influx of Ca2+ can lead to excitotoxicity. Excitotoxicity is implied to be involved in some neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease and Huntington's disease.[3][4][5][6] Blocking of NMDA receptors could therefore, in theory, be useful in treating such diseases.[3][4][5] It is, however, important to preserve physiological NMDA receptor activity while trying to block its excessive, excitotoxic activity. This can possibly be achieved by uncompetitive antagonists, blocking the receptors ion channel when excessively open.[5]
Uncompetitive NMDA receptor antagonists, or channel blockers, enter the channel of the NMDA receptor after it has been activated and thereby block the flow of ions.[1][3] MK-801, ketamine, amantadine and memantine are examples of such antagonists,[1] see figure 1. The off-rate of an antagonist from the receptors channel is an important factor as too slow off-rate can interfere with normal function of the receptor and too fast off-rate may give ineffective blockade of an excessively open receptor.[5]
Memantine is an example of an uncompetitive channel blocker of the NMDA receptor, with a relatively rapid off-rate and low affinity. At physiological pH its amine group is positively charged and its block of the channel is voltage-dependent.[5] It thereby mimics the physiological function of Mg2+ as channel blocker.[2] Memantine only blocks NMDA receptor associated channels during prolonged activation of the receptor, as it occurs under excitotoxic conditions, by replacing magnesium at the binding site. During normal receptor activity the channels only stay open for several milliseconds and under those circumstances memantine is unable to bind within the channels and therefore doesn't interfere with normal synaptic activity.[7]
Development of NMDA receptor antagonist
The main problem with the development of NMDA antagonist for neuroprotection is that the physiological NMDA receptor activity is essential for normal neuronal function. Complete blocking of all NMDA receptor activity therefore results in adverse side effects such as hallucination, agitation and anesthesia. To be clinically accepted the NMDA receptor antagonist must block excessive activation without blocking the normal function.[7] Figure 3 shows simplified models of various types of NMDA receptor antagonists, which will be discussed further.
Competitive NMDA receptor antagonists
Competitive NMDA receptor antagonists, which were developed first, are not a good option because they compete and bind to the same site (NR2 subunit) on the receptor as the agonist, glutamate, and therefore block normal function also.[7][19] They will block healthy areas of the brain prior to having an impact on pathological areas, because healthy areas contain lower levels of agonist than pathological areas. These antagonists can be displaced from the receptor by high concentration of glutamate which can exist under excitotoxic circumstances.[3]
Uncompetitive NMDA receptor antagonists

Uncompetitive NMDA receptor antagonists however block within the ion channel at the Mg2+ site (pore region) and in that way prevent excessive influx of Ca2+. Uncompetitive block refers to a type of block that increased concentration of glutamate cannot overcome and is dependent upon prior activation of the receptor by the agonist, i.e. it only enters the channel when it is opened by agonist.[7][20] The Mg2+ block itself is too transient and flickery and therefore doesn't block excessive Ca2+ influx to the extent necessary to prevent neurological toxicity.[7] High affinity antagonists for the Mg2+ site are on the other hand good excitotoxicity blockers, such as MK-801. They block open ion channels but the problem is when the ion channels close they get trapped inside resulting in undesirable side effects because of blocking normal as well as excessive activity.[7][20] MK-801 cannot be given to humans because of that long dwell time in the channels and its blocking causes drowsiness and even coma. It's therefore considered to be clinically unacceptable. Phencyclidine, which has slightly shorter dwell time but still to excessive, causes hallucination and is therefore not a good agent either for neurodegenerative diseases. Ketamine is another example of drug with slightly shorter dwell time but still excessive and it is used as anesthetic.[7] Chemical structures of MK-801, Phencyclidine and Ketamine can been seen in figure 4.
Memantine and related compounds

Because of these adverse side effects of high affinity blockers the search for clinically successful NMDA receptor antagonists for neurodegenerative diseases continued and focused on developing low affinity blockers. However the affinity could not be too low and dwell time not too short (as seen with Mg2+) where membrane depolarization relieves the block. The discovery was thereby development of uncompetitive antagonist with longer dwell time than Mg2+ in the channel but shorter than MK-801. That way the drug obtained would only block excessively open NMDA receptor associated channels but not normal neurotransmission.[7][20] Memantine is that drug. It is a derivative of amantadine which was first an anti-influenza agent but was later discovered by coincidence to have efficacy in Parkinson's disease. Chemical structures of memantine and amantadine can been seen in figure 5. The compound was first thought to be dopaminergic or anticholinergic but was later found to be an NMDA receptor antagonist.[2][7]
Memantine is the first drug approved for treatment of severe and more advanced Alzheimer's disease, which for example anticholinergic drugs do not do much good for.[20] It helps recovery of synaptic function and in that way improves impaired memory and learning.[6] In 2015 memantine is also in trials for therapeutic importance in additional neurological disorders.[21]
Many second-generation memantine derivatives have been in development that may show even better neuroprotective effects, where the main thought is to use other safe but effective modulatory sites on the NMDA receptor in addition to its associated ion channel.[21]
Neramexane

An example of memantine derivative is neramexane which was discovered by studying number of aminoalkyl cyclohexanes, with memantine as the template, as NMDA receptor antagonists. Neramexane, which can been seen in figure 6, binds to the same site as memantine within the NMDA receptor associated channel and with comparable affinity. It does also show very similar bioavailability and blocking kinetics in vivo as memantine. Neramexane went to clinical trials for four indications, including Alzheimer's disease.[12]
Nitromemantine
The NMDA receptor is regulated via nitrosylation and aminoadamantane can be used as a target-directed shuttle to bring nitrogen oxide (NO) close to the site within the NMDA receptor where it can nitrosylate and regulate the ion channel conductivity.[12] A NO donor that can be used to decrease NMDA receptor activity is the alkyl nitrate nitroglycerin. Unlike many other NO donors, alkyl nitrates do not have potential NO associated neurotoxic effects. Alkyl nitrates donate NO in the form of a nitro group as seen in figure 7, -NO2-, which is a safe donor that avoids neurotoxicity. The nitro group must be targeted to the NMDA receptor, otherwise other effects of NO such as dilatation of blood vessels and consequent hypotension could result.[21] Nitromemantine is a second-generation derivative of memantine, it reduces excitotoxicity mediated by overactivation of the glutamatergic system by blocking NMDA receptor without sacrificing safety. Provisional studies in animal models show that nitromemantines are more effective than memantine as neuroprotectants, both in vitro and in vivo. Memantine and newer derivatives could become very important weapons in the fight against neuronal damage.[5]

Structure activity relationship (SAR)

Memantine (1-amino-3,5-dimethyladamantane) is an aminoalkyl cyclohexane derivative and an atypical drug compound with non-planar, three dimensional tricyclic structure. Figure 8 shows SAR for aminoalkyl cyclohexane derivative. Memantine has several important features in its structure for its effectiveness:
- Three-ring structure with a bridgehead amine, -NH2
- The -NH2 group is protonated under physiological pH of the body to carry a positive charge, -NH3+
- Two methyl (CH3) side groups which serve to prolong the dwell time and increase stability as well as affinity for the NMDA receptor channel compared with amantadine (1-adamantanamine).[5][20]
Despite the small structural difference between memantine and amantadine, two adamantane derivatives, the affinity for the binding site of NR1/NR2B subunit is much greater for memantine. In patch-clamp measurements memantine has an IC50 of (2.3+0.3) µM while amantadine has an IC50 of (71.0+11.1) µM.[12] The binding site with the highest affinity is called the dominant binding site. It involves a connection between the amine group of memantine and the NR1-N161 binding pocket of the NR1/NR2B subunit. The methyl side groups play an important role in increasing the affinity to the open NMDA receptor channels and making it a much better neuroprotective drug than amantadine. The binding pockets for the methyl groups are considered to be at the NR1-A645 and NR2B-A644 of the NR1/NR2B.[16] The binding pockets are shown in figure 2. Memantine binds at or near to the Mg2+ site inside the NMDA receptor associated channel. The -NH2 group on memantine, which is protonated under physiological pH of the body, represents the region that binds at or near to the Mg2+ site.[5] Adding two methyl groups to the -N on the memantine structure has shown to decrease affinity, giving an IC50 value of (28.4+1.4) µM.[12]
Second generation derivative of memantine; Nitromemantine
Several derivatives of Nitromemantine, a second-generation derivative of memantine, have been synthesized in order to perform a detailed structure activity relationship (SAR) of these novel drugs. One class, containing a nitro (NO2) group opposite to the bridgehead amine (NH2), showed a promising outcome. Nitromemantine utilizes memantine binding site on the NMDA receptor to target the NOx (X= 1 or 2) group for interaction with the S- nitrosylation/redox site external to the memantine binding site. Lengthening the side chains of memantine compensates for the worse drug affinity in the channel associated with the addition of the –ONO2 group[22]
Therapeutic application
Excitotoxicity is implied to be involved in some neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis.[3][4][5][6] Blocking of NMDA receptors could therefore, in theory, be useful in treating such diseases.[3][4][5] It is, however, important to preserve physiological NMDA receptor activity while trying to block its excessive, excitotoxic activity. This can possibly be achieved by uncompetitive antagonists, blocking the receptors ion channel when excessively open [5]
Memantine is an example of uncompetitive NMDA receptor antagonist that has approved indication for the neurodegenerative disease Alzheimer's disease. In 2015 memantine is still in clinical trials for additional neurological diseases.[16][21]
References
- 1 2 3 4 Johnson, J.W. & Kotermanski, S.E. (2006). Mechanism of action of memantine. Current Opinion in Pharmacology, 6(1), 61-67.
- 1 2 3 4 Dominguez, Evangelyn; Chin, Ting-Yu; Chen, Chih-Ping; Wu, Tzong-Yuan (December 2011). "Management of moderate to severe Alzheimer's disease: Focus on memantine". Taiwanese Journal of Obstetrics and Gynecology. 50 (4): 415–423. doi:10.1016/j.tjog.2011.10.004.
- 1 2 3 4 5 6 7 8 Chen, Huei-Sheng Vincent; Lipton, Stuart A. (June 2006). "The chemical biology of clinically tolerated NMDA receptor antagonists". Journal of Neurochemistry. 97 (6): 1611–1626. doi:10.1111/j.1471-4159.2006.03991.x.
- 1 2 3 4 5 Kemp, J. A., & McKernan, R. M. (2002). NMDA receptor pathways as drug targets. Nature Neuroscience, 5(11), 1039–1042.
- 1 2 3 4 5 6 7 8 9 10 11 12 Lipton, S.A. (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nature Reviews Drug Discovery, 5(2), 160-170.
- 1 2 3 4 Koch, Horst; Szecsey, Alexander; Haen, Ekkehard (1 January 2004). "NMDA-antagonism (Memantine): An Alternative Pharmacological Therapeutic Principle in Alzheimers and Vascular Dementia". Current Pharmaceutical Design. 10 (3): 253–259. doi:10.2174/1381612043386392.
- 1 2 3 4 5 6 7 8 9 10 Lipton, Stuart A. (January 2004). "Failures and successes of NMDA receptor antagonists: Molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults". NeuroRX. 1 (1): 101–110. doi:10.1602/neurorx.1.1.101.
- ↑ Yamakura, T; Shimoji, K (October 1999). "Subunit- and site-specific pharmacology of the NMDA receptor channel". Progress in Neurobiology. 59 (3): 279–298. doi:10.1016/S0301-0082(99)00007-6.
- ↑ Watkins, Jeffrey C; Jane, David E (2 February 2009). "The glutamate story". British Journal of Pharmacology. 147 (S1): S100–S108. doi:10.1038/sj.bjp.0706444.
- ↑ Paoletti, Pierre; Neyton, Jacques (February 2007). "NMDA receptor subunits: function and pharmacology". Current Opinion in Pharmacology. 7 (1): 39–47. doi:10.1016/j.coph.2006.08.011.
- ↑ Dingledine, Raymond, Borges, Karin, Bowie, Derek, & Traynelis, Stephen F. (1999). The Glutamate Receptor Ion Channels. Pharmacological Reviews, 51(1), 7-62.
- 1 2 3 4 5 Wanka, Lukas; Iqbal, Khalid; Schreiner, Peter R. (8 May 2013). "The Lipophilic Bullet Hits the Targets: Medicinal Chemistry of Adamantane Derivatives". Chemical Reviews. 113 (5): 3516–3604. doi:10.1021/cr100264t.
- ↑ Furukawa, Hiroyasu; Singh, Satinder K; Mancusso, Romina; Gouaux, Eric (10 November 2005). "Subunit arrangement and function in NMDA receptors". Nature. 438 (7065): 185–192. doi:10.1038/nature04089.
- 1 2 Loftis, J. M., & Janowsky, A. (2003). The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther, 97(1), 55-85.
- 1 2 Kristiansen, Lars V; Huerta, Ibone; Beneyto, Monica; Meador-Woodruff, James H (February 2007). "NMDA receptors and schizophrenia". Current Opinion in Pharmacology. 7 (1): 48–55. doi:10.1016/j.coph.2006.08.013.
- 1 2 3 4 Limapichat, Walrati; Yu, Wesley Y.; Branigan, Emma; Lester, Henry A.; Dougherty, Dennis A. (20 February 2013). "Key Binding Interactions for Memantine in the NMDA Receptor". ACS Chemical Neuroscience. 4 (2): 255–260. doi:10.1021/cn300180a.
- ↑ Maher, T.J. (2013). Anesthetic agents: General and local anesthetics. In: T.L. Lemke & D.A. Williams (editors). Foye's Principles of Medicinal Chemistry. (Chapter 16). Philadelphia: Lippincott Williams & Wilkins
- ↑ Danysz, Wojciech; Parsons, Chris G. (September 2003). "The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer's disease: preclinical evidence". International Journal of Geriatric Psychiatry. 18 (S1): S23–S32. doi:10.1002/gps.938.
- ↑ Monaghan, Daniel T.; Jane, David E. (2009). "Pharmacology of NMDA Receptors". In Van Dongen, AM. Biology of the NMDA Receptor. Boca Raton, Florida: CRC Press. ISBN 978-1-4200-4414-0.
- 1 2 3 4 5 Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. (January 2005). "Dementia of Alzheimer's disease and other neurodegenerative disorders—memantine, a new hope". Pharmacological Research. 51 (1): 1–17. doi:10.1016/j.phrs.2004.05.005.
- 1 2 3 4 Lipton, Stuart A. (2007). Pathologically activated therapeutics for neuroprotection. Nature Reviews: Neuroscience, 8(10), 803-808
- ↑ Takahashi, Hiroto; Xia, Peng; Cui, Jiankun; Talantova, Maria; Bodhinathan, Karthik; Li, Wenjun; Holland, Emily A.; Tong, Gary; Piña-Crespo, Juan; Zhang, Dongxian; Nakanishi, Nobuki; Larrick, James W.; McKercher, Scott R.; Nakamura, Tomohiro; Wang, Yuqiang; Lipton, Stuart A. (19 October 2015). "Pharmacologically targeted NMDA receptor antagonism by NitroMemantine for cerebrovascular disease". Scientific Reports. 5: 14781. doi:10.1038/srep14781.