Nucleic acid sequence

A nucleic acid sequence is a succession of letters that indicate the order of nucleotides within a DNA (using GACT) or RNA (GACU) molecule. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
The sequence has capacity to represent information. Biological deoxyribonucleic acid represents the information which directs the functions of a living thing.
Nucleic acids also have a secondary structure and tertiary structure. Primary structure is sometimes mistakenly referred to as primary sequence. Conversely, there is no parallel concept of secondary or tertiary sequence.
Nucleotides
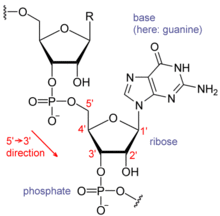
Nucleic acids consist of a chain of linked units called nucleotides. Each nucleotide consists of three subunits: a phosphate group and a sugar (ribose in the case of RNA, deoxyribose in DNA) make up the backbone of the nucleic acid strand, and attached to the sugar is one of a set of nucleobases. The nucleobases are important in base pairing of strands to form higher-level secondary and tertiary structure such as the famed double helix.
The possible letters are A, C, G, and T, representing the four nucleotide bases of a DNA strand — adenine, cytosine, guanine, thymine — covalently linked to a phosphodiester backbone. In the typical case, the sequences are printed abutting one another without gaps, as in the sequence AAAGTCTGAC, read left to right in the 5' to 3' direction. With regards to transcription, a sequence is on the coding strand if it has the same order as the transcribed RNA.
One sequence can be complementary to another sequence, meaning that they have the base on each position in the complementary (i.e. A to T, C to G) and in the reverse order. For example, the complementary sequence to TTAC is GTAA. If one strand of the double-stranded DNA is considered the sense strand, then the other strand, considered the antisense strand, will have the complementary sequence to the sense strand.
Notation
While A, T, C, and G represent a particular nucleotide at a position, there are also letters that represent ambiguity which are used when more than one kind of nucleotide could occur at that position. The rules of the International Union of Pure and Applied Chemistry (IUPAC) are as follows:[1]
- A = adenine
- C = cytosine
- G = guanine
- T = thymine
- R = G A (purine)
- Y = T C (pyrimidine)
- K = G T (keto)
- M = A C (amino)
- S = G C (strong bonds)
- W = A T (weak bonds)
- B = G T C (all but A)
- D = G A T (all but C)
- H = A C T (all but G)
- V = G C A (all but T)
- N = A G C T (any)
These symbols are also valid for RNA, except with U (uracil) replacing T (thymine).[1]
Apart from adenine (A), cytosine (C), guanine (G), thymine (T) and uracil (U), DNA and RNA also contain bases that have been modified after the nucleic acid chain has been formed. In DNA, the most common modified base is 5-methylcytidine (m5C). In RNA, there are many modified bases, including pseudouridine (Ψ), dihydrouridine (D), inosine (I), ribothymidine (rT) and 7-methylguanosine (m7G).[2][3] Hypoxanthine and xanthine are two of the many bases created through mutagen presence, both of them through deamination (replacement of the amine-group with a carbonyl-group). Hypoxanthine is produced from adenine, xanthine from guanine.[4] Similarly, deamination of cytosine results in uracil.
Biological significance

In biological systems, nucleic acids contain information which is used by a living cell to construct specific proteins. The sequence of nucleobases on a nucleic acid strand is translated by cell machinery into a sequence of amino acids making up a protein strand. Each group of three bases, called a codon, corresponds to a single amino acid, and there is a specific genetic code by which each possible combination of three bases corresponds to a specific amino acid.
The central dogma of molecular biology outlines the mechanism by which proteins are constructed using information contained in nucleic acids. DNA is transcribed into mRNA molecules, which travels to the ribosome where the mRNA is used as a template for the construction of the protein strand. Since nucleic acids can bind to molecules with complementary sequences, there is a distinction between "sense" sequences which code for proteins, and the complementary "antisense" sequence which is by itself nonfunctional, but can bind to the sense strand.
Sequence determination

DNA sequencing is the process of determining the nucleotide sequence of a given DNA fragment. The sequence of the DNA of a living thing encodes the necessary information for that living thing to survive and reproduce. Therefore, determining the sequence is useful in fundamental research into why and how organisms live, as well as in applied subjects. Because of the importance of DNA to living things, knowledge of a DNA sequence may be useful in practically any biological research. For example, in medicine it can be used to identify, diagnose and potentially develop treatments for genetic diseases. Similarly, research into pathogens may lead to treatments for contagious diseases. Biotechnology is a burgeoning discipline, with the potential for many useful products and services.
RNA is not sequenced directly. Instead, it is copied to a DNA by reverse transcriptase, and this DNA is then sequenced.
Current sequencing methods rely on the discriminatory ability of DNA polymerases, and therefore can only distinguish four bases. An inosine (created from adenosine during RNA editing) is read as a G, and 5-methyl-cytosine (created from cytosine by DNA methylation) is read as a C. With current technology, it is difficult to sequence small amounts of DNA, as the signal is too weak to measure. This is overcome by polymerase chain reaction (PCR) amplification.
Digital representation

Once a nucleic acid sequence has been obtained from an organism, it is stored in silico in digital format. Digital genetic sequences may be stored in sequence databases, be analyzed (see Sequence analysis below), be digitally altered and be used as templates for creating new actual DNA using artificial gene synthesis.
Sequence analysis
Digital genetic sequences may be analyzed using the tools of bioinformatics to attempt to determine its function.
Genetic testing
The DNA in an organism's genome can be analyzed to diagnose vulnerabilities to inherited diseases, and can also be used to determine a child's paternity (genetic father) or a person's ancestry. Normally, every person carries two variations of every gene, one inherited from their mother, the other inherited from their father. The human genome is believed to contain around 20,000 - 25,000 genes. In addition to studying chromosomes to the level of individual genes, genetic testing in a broader sense includes biochemical tests for the possible presence of genetic diseases, or mutant forms of genes associated with increased risk of developing genetic disorders.
Genetic testing identifies changes in chromosomes, genes, or proteins.[5] Usually, testing is used to find changes that are associated with inherited disorders. The results of a genetic test can confirm or rule out a suspected genetic condition or help determine a person's chance of developing or passing on a genetic disorder. Several hundred genetic tests are currently in use, and more are being developed.[6][7]
Sequence alignment
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be due to functional, structural, or evolutionary relationships between the sequences.[8] If two sequences in an alignment share a common ancestor, mismatches can be interpreted as point mutations and gaps as insertion or deletion mutations (indels) introduced in one or both lineages in the time since they diverged from one another. In sequence alignments of proteins, the degree of similarity between amino acids occupying a particular position in the sequence can be interpreted as a rough measure of how conserved a particular region or sequence motif is among lineages. The absence of substitutions, or the presence of only very conservative substitutions (that is, the substitution of amino acids whose side chains have similar biochemical properties) in a particular region of the sequence, suggest[9] that this region has structural or functional importance. Although DNA and RNA nucleotide bases are more similar to each other than are amino acids, the conservation of base pairs can indicate a similar functional or structural role.[10]
Computational phylogenetics makes extensive use of sequence alignments in the construction and interpretation of phylogenetic trees, which are used to classify the evolutionary relationships between homologous genes represented in the genomes of divergent species. The degree to which sequences in a query set differ is qualitatively related to the sequences' evolutionary distance from one another. Roughly speaking, high sequence identity suggests that the sequences in question have a comparatively young most recent common ancestor, while low identity suggests that the divergence is more ancient. This approximation, which reflects the "molecular clock" hypothesis that a roughly constant rate of evolutionary change can be used to extrapolate the elapsed time since two genes first diverged (that is, the coalescence time), assumes that the effects of mutation and selection are constant across sequence lineages. Therefore, it does not account for possible difference among organisms or species in the rates of DNA repair or the possible functional conservation of specific regions in a sequence. (In the case of nucleotide sequences, the molecular clock hypothesis in its most basic form also discounts the difference in acceptance rates between silent mutations that do not alter the meaning of a given codon and other mutations that result in a different amino acid being incorporated into the protein.) More statistically accurate methods allow the evolutionary rate on each branch of the phylogenetic tree to vary, thus producing better estimates of coalescence times for genes.
Sequence motifs
Frequently the primary structure encodes motifs that are of functional importance. Some examples of sequence motifs are: the C/D[11] and H/ACA boxes[12] of snoRNAs, Sm binding site found in spliceosomal RNAs such as U1, U2, U4, U5, U6, U12 and U3, the Shine-Dalgarno sequence,[13] the Kozak consensus sequence[14] and the RNA polymerase III terminator.[15]
Long range correlations
Peng [16][17] found the existence of long-range correlations in the non-coding base pair sequences of DNA. In contrast, such correlations seem not to appear in coding DNA sequences.
Sequence entropy
In Bioinformatics, a sequence entropy, also known as sequence complexity or information profile,[18] is a numerical sequence providing a quantitative measure of the local complexity of a DNA sequence, independently of the direction of processing. The manipulations of the information profiles enable the analysis of the sequences using alignment-free techniques, such as for example in motif and rearrangements detection.[18][19] [20]
See also
References
- 1 2 Nomenclature for Incompletely Specified Bases in Nucleic Acid Sequences, NC-IUB, 1984.
- ↑ "BIOL2060: Translation". mun.ca.
- ↑ "Research". uw.edu.pl.
- ↑ T Nguyen, D Brunson, C L Crespi, B W Penman, J S Wishnok, and S R Tannenbaum, DNA damage and mutation in human cells exposed to nitric oxide in vitro, Proc Natl Acad Sci U S A. 1992 April 1; 89(7): 3030–3034
- ↑ "What is genetic testing?". Genetics Home Reference. 16 March 2015.
- ↑ "Genetic Testing". nih.gov.
- ↑ "Definitions of Genetic Testing". Definitions of Genetic Testing (Jorge Sequeiros and Bárbara Guimarães). EuroGentest Network of Excellence Project. 2008-09-11. Archived from the original on February 4, 2009. Retrieved 2008-08-10.
- ↑ Mount DM. (2004). Bioinformatics: Sequence and Genome Analysis (2nd ed.). Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY. ISBN 0-87969-608-7.
- ↑ Ng, P. C.; Henikoff, S. (2001). "Predicting Deleterious Amino Acid Substitutions". Genome Research. 11 (5): 863–874. doi:10.1101/gr.176601. PMC 311071
 . PMID 11337480.
. PMID 11337480. - ↑ Witzany, G (2016) Crucial steps to life: From chemical reactions to code using agents. Biosystems 140: 49-57.
- ↑ Samarsky, DA; Fournier MJ; Singer RH; Bertrand E (1998). "The snoRNA box C/D motif directs nucleolar targeting and also couples snoRNA synthesis and localization". The EMBO Journal. 17 (13): 3747–3757. doi:10.1093/emboj/17.13.3747. PMC 1170710. PMID 9649444.
- ↑ Ganot, Philippe; Caizergues-Ferrer, Michèle; Kiss, Tamás (1 April 1997). "The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation". Genes & Development. 11 (7): 941–956. doi:10.1101/gad.11.7.941. PMID 9106664.
- ↑ Shine J, Dalgarno L (1975). "Determinant of cistron specificity in bacterial ribosomes". Nature. 254 (5495): 34–8. doi:10.1038/254034a0. PMID 803646.
- ↑ Kozak M (October 1987). "An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs". Nucleic Acids Res. 15 (20): 8125–8148. doi:10.1093/nar/15.20.8125. PMC 306349. PMID 3313277.
- ↑ Bogenhagen DF, Brown DD (1981). "Nucleotide sequences in Xenopus 5S DNA required for transcription termination.". Cell. 24 (1): 261–70. doi:10.1016/0092-8674(81)90522-5. PMID 6263489.
- ↑ Peng, C.-K.; Buldyrev, S. V.; Goldberger, A. L.; Havlin, S.; Sciortino, F.; Simons, M.; Stanley, H. E. (1992). "Long-range correlations in nucleotide sequences". Nature. 356 (6365): 168–170. doi:10.1038/356168a0. ISSN 0028-0836.
- ↑ Peng, C.-K.; Buldyrev, S. V.; Havlin, S.; Simons, M.; Stanley, H. E.; Goldberger, A. L. (1994). "Mosaic organization of DNA nucleotides". Physical Review E. 49 (2): 1685–1689. doi:10.1103/PhysRevE.49.1685. ISSN 1063-651X.
- 1 2 Pinho, A; Garcia, S; Pratas, D; Ferreira, P (Nov 21, 2013). "DNA Sequences at a Glance.". PLOS ONE. 8 (11): e79922. doi:10.1371/journal.pone.0079922. PMC 3836782. PMID 24278218.
- ↑ Pratas, D; Silva, R; Pinho, A; Ferreira, P (May 18, 2015). "An alignment-free method to find and visualise rearrangements between pairs of DNA sequences.". Scientific Reports (Group Nature). 5 (10203): 10203. doi:10.1038/srep10203. PMID 25984837.
- ↑ Troyanskaya, O; Arbell, O; Koren, Y; Landau, G; Bolshoy, A (2002). "Sequence complexity profiles of prokaryotic genomic sequences: A fast algorithm for calculating linguistic complexity.". Bioinformatics. 18 (5): 679–88. doi:10.1093/bioinformatics/18.5.679. PMID 12050064.
External links
| Wikimedia Commons has media related to Nucleic acid sequence. |