CDKN2A
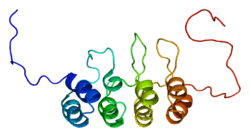


| View/Edit Human | View/Edit Mouse |
CDKN2A, also known as cyclin-dependent kinase Inhibitor 2A, is a gene which in humans is located at chromosome 9, band p21.3.[3] It is ubiquitously expressed in many tissues and cell types.[4] The gene codes for two proteins, including p16 (or p16INK4a) and p14arf.[5] Both act as tumor suppressors by regulating the cell cycle. p16 inhibits cyclin dependent kinases 4 and 6 (CDK4 and CDK6) and thereby activates the retinoblastoma (Rb) family of proteins, which block traversal from G1 to S-phase. p14ARF (also known as p19ARF in the mouse) activates the p53 tumor suppressor. Somatic mutations of CDKN2A are common in the majority of human cancers, with estimates that CDKN2a is the second most commonly inactivated gene in cancer after p53. Germline mutations of CDKN2a are associated with familial melanoma, glioblastoma and pancreatic cancer.[6] The CDKN2A gene also contains one of 27 SNPs associated with increased risk of coronary artery disease.[7]
Structure[8]
Gene
The CDKN2A gene resides on chromosome 9 at the band 9p21 and contains 8 exons.[9] This gene encodes two proteins, p16 and ARF, which are transcribed from the same second and third exons but alternative first exons: p16 from exon 1α and ARF from exon 1β. As a result, they are translated from different reading frames and therefore possess completely different amino acid sequences.[10] In addition to p16 and ARF, this gene produces 4 other isoforms through alternative splicing.[11]
Protein
p16
This protein belongs to the CDKN2 cyclin-dependent kinase inhibitor family.[11] p16 comprises four ankyrin repeats, each spanning a length of 33 amino acid residues and, in the tertiary structure, forming a helix-turn-helix motif. One exception is the second ankyrin repeat, which contains only one helical turn. These four motifs are connected by three loops such that they are oriented perpendicular to the helical axes.
According to its solvent-accessible surface representation, p16 features clustered charged groups on its surface and a pocket located on the right side with a negatively charged left inner wall and a positively charged right inner wall.[12]
ARF
The size of this protein is 14 kDa in humans.[13] Within the N-terminal half of ARF are highly hydrophobic domains that serve as mitochondrial import sequences.
Function
P14ARF
P14ARF has become a central actor of the cell cycle regulation process as it participates to the ARF-MDM2-p53 pathway and the Rb-E2F-1 pathway.[14] It is the physiological inhibitor of MDM2, an E3 ubiquitin ligase controlling the activity and stability of P53, and loss of P14ARF activity may have a similar effect as loss of P53.[15] P14ARF induces cell cycle arrest in G2 phase and subsequent apoptosis in a P53-dependent and P53-independent manner, and thus is regarded as a tumor suppressor.[16][17][18][19] In addition, P14ARF could down-regulate E2F-dependent transcription and plays a role in the control of the G1 to S phase transition as well.[20]
P16(INK4A)
P16 interacts with Rb and controls the G1 to S transition. It binds to CDK4/6 inhibiting its kinase activity and prevents Rb phosphorylation. Therefore, Rb remains associated with transcription factor E2F1, preventing transcription of E2F1 target genes which are crucial for the G1/S transition. During this process, a feedback loop exists between P16 and Rb, and P16 expression is controlled by Rb.[21][22] P16/Rb pathway collaborates with the mitogenic signaling cascade for the induction of reactive oxygen species, which activates the protein kinase C delta, leading to an irreversible cell cycle arrest. Thus P16 participates not only in the initiation but also in the maintenance of cellular senescence, as well in tumor suppression.[23][24] On the other hand, some specific tumors harbor high levels of P16, and its function in limitation of tumorigenic progression has been inactivated via the loss of Rb.[24][25]
Clinical relevance
In human cancer cell lines derived from various tumor types, a high frequency of genetic and epigenetic alterations (e.g., promoter hyper-methylation, homozygous deletion or mutation) in the CDKN2A gene has been observed. Accordingly, epigenetic/genetic modulation of changes in CDKN2A might be a promising strategy for prevention or therapy of cancer.
The CDKN2A gene is located on the chromosome 9p21 locus, which is intriguing for several reasons. First, this region is well known in cancer genetics as one of the most common sites of deletions leading to hereditary forms of cutaneous malignant melanoma.[10][26] Second, genome wide association studies have reported a significant association of chromosome 9p21 with coronary artery disease and myocardial infarction[27] as well as the progression of atherosclerosis.[28]
Furthermore, changes in CDKN2A status are highly variable depending on the type of cancer. In addition to skin cancer such as melanoma, changes of CDKN2A have been described in a wide spectrum of cancer types such as gastric lymphoma,[29] Burkitt’s lymphoma,[30] head & neck squamous cell carcinoma,[31] oral cancer,[32] pancreatic adenocarcinoma,[33] non-small cell lung carcinoma,[34] esophageal squamous cell carcinoma,[35] gastric cancer,[36] colorectal cancer,[37] epithelial ovarian carcinoma[38] and prostate cancer[39]
Familial melanoma
CDKN2A is made up of four sections of exons – exon 1β, exon 1α,exon 2, and exon 3. These exons are used to create two proteins named p16 and p14ARF. Protein p16, created by exon 1α and exon 2, is responsible for tumor creation of genetic melanoma. When working normally, p16 binds to the cyclic dependent kinases CDK4 to inhibit their ability to create tumors, but when inactivated the suppression no longer occurs.[40] When a mutation occurs in protein p16, it prevents the protein kinase of CDK4, which results in the inactivation of the tumor suppressor gene.[40] Thus, kick starting the development of melanoma.
Melanoma only occurs in a small proportion of the population. Only 10% of those who have melanoma acquired it genetically.[41] This disease is an autosomal dominant gene.[40] If only two family members have melanoma, there is a 5% chance somebody in the next generation will acquire the mutated gene. Also, there is a 20-40% chance of getting hereditary melanoma in a family if 3 or more people in the past generation had melanoma. For those who carry the hereditary mutated gene CDKN2A, acquiring skin cancer is a lot easier.[41] Those who have the gene are far more likely to get melanoma a second or third time compared to those who don’t genetically have this gene.[42] The population that is affected by this mutation has a high familial history of melanoma or atypical moles and birth marks in large numbers, a history of primary melanoma/cancers in general, immunosuppression, skin that burns easily and doesn't tan, freckling, blue eyes, red hair, or a history of blistering.[41] People with these high risk factors are more likely to carry inherited mutations in CDKN2A.[42] For those who have a gene mutation, the severity is also dependent on the environmental surroundings. Out of those who carry the gene, those who express the phenotype and actually developed melanoma have a history of more sun exposure, and light skin compared to those who also had the gene but never actually developed melanoma.[42] This suggests that this gene co-works with ones surrounding environment. If two individuals are selected who carry the CDKN2A mutation, and both genetically have the same probability of acquiring skin cancer, but one is from Australia and the other is from Europe, there is a 58% the European will acquire cancer compared to a 91% chance the Australian will get it.[42] This is because the factors mentioned earlier pertaining to those who are more susceptible to the disease and also dependent on the amount of sunscreen one wears and the UV radiation potency in their environment.[41]
Clinical marker
A multi-locus genetic risk score study based on a combination of 27 loci, including the CDKN2A gene, identified individuals at increased risk for both incident and recurrent coronary artery disease events, as well as an enhanced clinical benefit from statin therapy. The study was based on a community cohort study (the Malmo Diet and Cancer study) and four additional randomized controlled trials of primary prevention cohorts (JUPITER and ASCOT) and secondary prevention cohorts (CARE and PROVE IT-TIMI 22).[7]
Therapeutic potential of BRAF kinase inhibitors
RAS-RAF-MEK-ERK MAP kinase pathway plays an important role in melanocytes, where melanoma arises.[43] Stimulation of membrane bound receptors including tyrosine kinases and G-proteins receptors promote activation of RAS then activates Raf kinases which in turn promotes MEK then ERK activation. All of these proteins work together to help cell survival. A BRAF mutation in this activation chain triggers maligant transformation in melanoma cells.[44] Melanoma relies on this BRAF mutation to grow and multiply as a tumor cell.
RNA interference of BRAF stops the multiplication of melanoma tumor cells.[44] This suggests that BRAF inhibitors such as sorafenib, PLX4720, PLX4032, GDC-0879,GSK2118436, or AZ628 may be affective in treating melanoma.[43] These have been tested and proven to slow down the production of melanoma due to BRAF mutation but PLX4032 and GSK2118436 show the most promise because they worked the most aggressively in early stages of cell development.[44] Although this drug helps slow down or shrink the tumors, there are many serious side affects to using these drugs that were found in the trials including arthralgia, rash, nausea, phosphosensitivity], fatigue, pruritus and palmo-plantar dysesthesia.[44]
Aging
Activation of the CDKN2a locus promotes the cellular senescence tumor suppressor mechanism, which is a permanent form of growth arrest. As senescent cells accumulate with aging, expression of CDKN2a increases exponentially with aging in all mammalian species tested to date, and has been argued to serve as a biomarker of physiological age.[45]
References
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- ↑ "CDKN2A". Genetics Home Reference. National Library of Medicine. January 2015. Retrieved April 14, 2015.
- ↑ "BioGPS - your Gene Portal System". biogps.org. Retrieved 2016-10-11.
- ↑ "Cyclin-Dependent Kinase Inhibitor 2A". GeneCards. Weizmann Institute of Science. Retrieved April 14, 2015.
- ↑ "Genetics of Skin Cancer". National Cancer Institute. Retrieved April 14, 2015.
- 1 2 Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield MJ, Devlin JJ, Nordio F, Hyde CL, Cannon CP, Sacks FM, Poulter NR, Sever PS, Ridker PM, Braunwald E, Melander O, Kathiresan S, Sabatine MS (June 2015). "Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials". Lancet. 385 (9984): 2264–71. doi:10.1016/S0140-6736(14)61730-X. PMID 25748612.
- ↑ Irvine M, Philipsz S, Frausto M, Mijatov B, Gallagher SJ, Fung C, Becker TM, Kefford RF, Rizos H (February 2010). "Amino terminal hydrophobic import signals target the p14(ARF) tumor suppressor to the mitochondria". Cell Cycle. 9 (4): 829–39. doi:10.4161/cc.9.4.10785. PMID 20107316.
- ↑ "CDKN2A cyclin dependent kinase inhibitor 2A [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2016-10-11.
- 1 2 Aoude LG, Wadt KA, Pritchard AL, Hayward NK (March 2015). "Genetics of familial melanoma: 20 years after CDKN2A". Pigment Cell & Melanoma Research. 28 (2): 148–60. doi:10.1111/pcmr.12333. PMID 25431349.
- 1 2 "CDKN2A - Cyclin-dependent kinase inhibitor 2A - Homo sapiens (Human) - CDKN2A gene & protein". www.uniprot.org. Retrieved 2016-10-11.
- ↑ Byeon, I. J.; Li, J.; Ericson, K.; Selby, T. L.; Tevelev, A.; Kim, H. J.; O'Maille, P.; Tsai, M. D. (1998-02-01). "Tumor suppressor p16INK4A: determination of solution structure and analyses of its interaction with cyclin-dependent kinase 4". Molecular Cell. 1 (3): 421–431. ISSN 1097-2765. PMID 9660926.
- ↑ Lo D, Zhang Y, Dai MS, Sun XX, Zeng SX, Lu H (March 2015). "Nucleostemin stabilizes ARF by inhibiting the ubiquitin ligase ULF". Oncogene. 34 (13): 1688–97. doi:10.1038/onc.2014.103. PMID 24769896.
- ↑ Karayan L, Riou JF, Séité P, Migeon J, Cantereau A, Larsen CJ (February 2001). "Human ARF protein interacts with topoisomerase I and stimulates its activity". Oncogene. 20 (7): 836–48. doi:10.1038/sj.onc.1204170. PMID 11314011.
- ↑ Kanellou P, Zaravinos A, Zioga M, Spandidos DA (June 2009). "Deregulation of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in basal cell carcinoma". The British Journal of Dermatology. 160 (6): 1215–21. doi:10.1111/j.1365-2133.2009.09079.x. PMID 19298278.
- ↑ Huang Y, Tyler T, Saadatmandi N, Lee C, Borgstrom P, Gjerset RA (July 2003). "Enhanced tumor suppression by a p14ARF/p53 bicistronic adenovirus through increased p53 protein translation and stability". Cancer Research. 63 (13): 3646–53. PMID 12839954.
- ↑ Chen D, Kon N, Li M, Zhang W, Qin J, Gu W (July 2005). "ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor". Cell. 121 (7): 1071–83. doi:10.1016/j.cell.2005.03.037. PMID 15989956.
- ↑ Miao L, Song Z, Jin L, Zhu YM, Wen LP, Wu M (February 2010). "ARF antagonizes the ability of Miz-1 to inhibit p53-mediated transactivation". Oncogene. 29 (5): 711–22. doi:10.1038/onc.2009.372. PMID 19901969.
- ↑ Eymin B, Leduc C, Coll JL, Brambilla E, Gazzeri S (March 2003). "p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice". Oncogene. 22 (12): 1822–35. doi:10.1038/sj.onc.1206303. PMID 12660818.
- ↑ Mason SL, Loughran O, La Thangue NB (June 2002). "p14(ARF) regulates E2F activity". Oncogene. 21 (27): 4220–30. doi:10.1038/sj.onc.1205524. PMID 12082609.
- ↑ Rayess H, Wang MB, Srivatsan ES (April 2012). "Cellular senescence and tumor suppressor gene p16". International Journal of Cancer. 130 (8): 1715–25. doi:10.1002/ijc.27316. PMC 3288293
 . PMID 22025288.
. PMID 22025288. - ↑ Li Y, Nichols MA, Shay JW, Xiong Y (December 1994). "Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb". Cancer Research. 54 (23): 6078–82. PMID 7954450.
- ↑ Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E (November 2006). "Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence". Nature Cell Biology. 8 (11): 1291–7. doi:10.1038/ncb1491. PMID 17028578.
- 1 2 Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES (August 2011). "The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments". Cell Cycle. 10 (15): 2497–503. doi:10.4161/cc.10.15.16776. PMC 3685613. PMID 21775818.
- ↑ Kelley MJ, Nakagawa K, Steinberg SM, Mulshine JL, Kamb A, Johnson BE (May 1995). "Differential inactivation of CDKN2 and Rb protein in non-small-cell and small-cell lung cancer cell lines". Journal of the National Cancer Institute. 87 (10): 756–61. PMID 7563154.
- ↑ Hayward NK (May 2003). "Genetics of melanoma predisposition". Oncogene. 22 (20): 3053–62. doi:10.1038/sj.onc.1206445. PMID 12789280.
- ↑ McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC (June 2007). "A common allele on chromosome 9 associated with coronary heart disease". Science. 316 (5830): 1488–91. doi:10.1126/science.1142447. PMID 17478681.
- ↑ Ye S, Willeit J, Kronenberg F, Xu Q, Kiechl S (July 2008). "Association of genetic variation on chromosome 9p21 with susceptibility and progression of atherosclerosis: a population-based, prospective study". Journal of the American College of Cardiology. 52 (5): 378–84. doi:10.1016/j.jacc.2007.11.087. PMID 18652946.
- ↑ Huang Q, Su X, Ai L, Li M, Fan CY, Weiss LM (October 2007). "Promoter hypermethylation of multiple genes in gastric lymphoma". Leukemia & Lymphoma. 48 (10): 1988–96. doi:10.1080/10428190701573224. PMID 17852707.
- ↑ Robaina MC, Faccion RS, Arruda VO, de Rezende LM, Vasconcelos GM, Apa AG, Bacchi CE, Klumb CE (February 2015). "Quantitative analysis of CDKN2A methylation, mRNA, and p16(INK4a) protein expression in children and adolescents with Burkitt lymphoma: biological and clinical implications". Leukemia Research. 39 (2): 248–56. doi:10.1016/j.leukres.2014.11.023. PMID 25542698.
- ↑ El-Naggar, A. K.; Lai, S.; Clayman, G.; Lee, J. K.; Luna, M. A.; Goepfert, H.; Batsakis, J. G. (1997-12-01). "Methylation, a major mechanism of p16/CDKN2 gene inactivation in head and neck squamous carcinoma". The American Journal of Pathology. 151 (6): 1767–1774. ISSN 0002-9440.
- ↑ Asokan GS, Jeelani S, Gnanasundaram N (October 2014). "Promoter hypermethylation profile of tumour suppressor genes in oral leukoplakia and oral squamous cell carcinoma". Journal of Clinical and Diagnostic Research. 8 (10): ZC09–12. doi:10.7860/JCDR/2014/9251.4949. PMID 25478438.
- ↑ Jiao L, Zhu J, Hassan MM, Evans DB, Abbruzzese JL, Li D (January 2007). "K-ras mutation and p16 and preproenkephalin promoter hypermethylation in plasma DNA of pancreatic cancer patients: in relation to cigarette smoking". Pancreas. 34 (1): 55–62. doi:10.1097/01.mpa.0000246665.68869.d4. PMID 17198183.
- ↑ Marchetti, A.; Buttitta, F.; Pellegrini, S.; Bertacca, G.; Chella, A.; Carnicelli, V.; Tognoni, V.; Filardo, A.; Angeletti, C. A. (1997-02-01). "Alterations of P16 (MTS1) in node-positive non-small cell lung carcinomas". The Journal of Pathology. 181 (2): 178–182. doi:10.1002/(SICI)1096-9896(199702)181:23.0.CO;2-5. ISSN 0022-3417.
- ↑ Qureshi MA, Jan N, Dar NA, Hussain M, Andrabi KI (September 2012). "A novel p16(INK4A) mutation associated with esophageal squamous cell carcinoma in a high risk population". Biomarkers. 17 (6): 552–6. doi:10.3109/1354750X.2012.699556. PMID 22724384.
- ↑ He D, Zhang YW, Zhang NN, Zhou L, Chen JN, Jiang Y, Shao CK (April 2015). "Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr virus-associated gastric carcinomas". Medical Oncology. 32 (4): 92. doi:10.1007/s12032-015-0525-y. PMID 25720522.
- ↑ Rajendran P, Dashwood WM, Li L, Kang Y, Kim E, Johnson G, Fischer KA, Löhr CV, Williams DE, Ho E, Yamamoto M, Lieberman DA, Dashwood RH (2015-01-01). "Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon". Clinical Epigenetics. 7: 102. doi:10.1186/s13148-015-0132-y. PMID 26388957.
- ↑ Bhagat R, Kumar SS, Vaderhobli S, Premalata CS, Pallavi VR, Ramesh G, Krishnamoorthy L (September 2014). "Epigenetic alteration of p16 and retinoic acid receptor beta genes in the development of epithelial ovarian carcinoma". Tumour Biology. 35 (9): 9069–78. doi:10.1007/s13277-014-2136-1. PMID 24913706.
- ↑ Ameri A, Alidoosti A, Hosseini SY, Parvin M, Emranpour MH, Taslimi F, Salehi E, Fadavip P (December 2011). "Prognostic Value of Promoter Hypermethylation of Retinoic Acid Receptor Beta (RARB) and CDKN2 (p16/MTS1) in Prostate Cancer". Chinese Journal of Cancer Research = Chung-Kuo Yen Cheng Yen Chiu. 23 (4): 306–11. doi:10.1007/s11670-011-0306-x. PMID 23358881.
- 1 2 3 Tsao H, Niendorf K (November 2004). "Genetic testing in hereditary melanoma". Journal of the American Academy of Dermatology. 51 (5): 803–8. doi:10.1016/j.jaad.2004.04.045. PMID 15523363.
- 1 2 3 4 Kefford R, Bishop JN, Tucker M, Bressac-de Paillerets B, Bianchi-Scarrá G, Bergman W, Goldstein A, Puig S, Mackie R, Elder D, Hansson J, Hayward N, Hogg D, Olsson H (November 2002). "Genetic testing for melanoma". The Lancet. Oncology. 3 (11): 653–4. doi:10.1016/s1470-2045(02)00894-x. PMID 12424065.
- 1 2 3 4 Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B, Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland EA, Mann GJ, Mantelli M, Nancarrow D, Platz A, Tucker MA (June 2002). "Geographical variation in the penetrance of CDKN2A mutations for melanoma". Journal of the National Cancer Institute. 94 (12): 894–903. doi:10.1093/jnci/94.12.894. PMID 12072543.
- 1 2 Pérez-Lorenzo R, Zheng B (February 2012). "Targeted inhibition of BRAF kinase: opportunities and challenges for therapeutics in melanoma". Bioscience Reports. 32 (1): 25–33. doi:10.1042/BSR20110068. PMC 3837566. PMID 21981139.
- 1 2 3 4 Solit DB, Rosen N (February 2011). "Resistance to BRAF inhibition in melanomas". The New England Journal of Medicine. 364 (8): 772–4. doi:10.1056/NEJMcibr1013704. PMID 21345109.
- ↑ Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE (November 2004). "Ink4a/Arf expression is a biomarker of aging". The Journal of Clinical Investigation. 114 (9): 1299–307. doi:10.1172/JCI22475. PMC 524230. PMID 15520862.