Anomeric effect

In organic chemistry, the anomeric effect or Edward-Lemieux effect is a stereoelectronic effect that describes the tendency of heteroatomic substituents adjacent to a heteroatom within a cyclohexane ring to prefer the axial orientation instead of the less hindered equatorial orientation that would be expected from steric considerations.[1] This effect was originally observed in pyranose rings by J. T. Edward in 1955 when studying carbohydrate chemistry.
The term "anomeric effect" was introduced in 1958.[2] The name comes from the term used to designate the C-1 carbon of a pyranose, the anomeric carbon. Isomers that differ only in the configuration at the anomeric carbon are called anomers. The anomers of glucopyranose are diastereomers, with the beta anomer having an OH group pointing up equatorially, and the alpha anomer having that OH group pointing down axially.
The anomeric effect can also be generalized to any cyclohexyl or linear system with the general formula C-Y-C-X, where Y is a heteroatom with one or more lone pairs, and X is an electronegative atom or group.[3] The magnitude of the anomeric effect is estimated at about 1–2 kcal/mol in the case of sugars, but is different for every molecule.
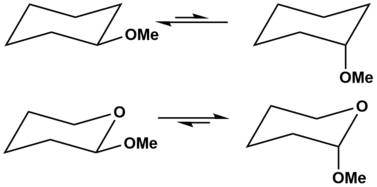
In the above case, the methoxy group on the cyclohexane ring (top) prefers the equatorial position. However, in the tetrahydropyran ring (bottom), the methoxy group prefers the axial position. This is because in the cyclohexane ring, Y= carbon, which is not a heteroatom, so the anomeric effect is not observed and sterics dominates the observed substituent position. In the tetrahydropyran ring, Y= oxygen, which is a heteroatom, so the anomeric effect contributes and stabilizes the observed substituent position. In both cases, X= OMe.
The anomeric effect is most often observed when Y= oxygen, but can also be seen with other lone pair bearing heteroatoms in the ring, such as nitrogen, sulfur, and phosphorus.[4] The exact method by which the anomeric effect causes stabilization is a point of controversy, and several hypotheses have been proposed to explain it.
Physical Explanation and Controversy
The physical reason for the anomeric effect is not completely understood. Several, in part conflicting, explanations have been offered and the topic is still not settled
Hyperconjugation
Cyclic Molecules
A widely accepted explanation is that there is a stabilizing interaction (hyperconjugation) between the unshared electron pair on the heteroatom (the endocyclic one in a sugar ring) and the σ* orbital for the axial (exocyclic) C–X bond. This causes the molecule to align the donating lone pair of electrons antiperiplanar (180°) to the σ* orbital lowering the overall energy of the system and causing more stability.[5]

Some authors also question the validity of this hyperconjugation model based on results from the quantum theory of atoms in molecules.[6] While most studies on the anomeric effects have been theoretical in nature, the n–σ* (hyperconjugation) hypothesis has also been extensively criticized on the basis that the electron density redistribution in acetals proposed by this hypothesis is not congruent with the known experimental chemistry of acetals and, in particular, the chemistry of monosaccharides.[7][8]
Acyclic Molecules
Hyperconjugation is also found in acyclic molecules containing heteroatoms, another form of the anomeric effect. If a molecule has an atom with a lone pair of electrons and the adjacent atom is able to accept electrons into the σ* orbital, hyperconjugation occurs, stabilizing the molecule. This forms a "no bond" resonance form. For this orbital overlap to occur, the trans, trans conformation is preferred for most heteroatoms, however for the stabilization to occur in dimethoxymethane, the gauche, gauche conformation is about 3–5 kcal/mol lower in energy (more stable) than the trans,trans conformation—this is about two times as big as the effect in sugars because there are two rotatable bonds(hence it is trans around both bonds or gauche around both) that are affected.[9]

Dipole Minimization
Another accepted explanation for the anomeric effect is the equatorial configuration has the dipoles involving both heteroatoms partially aligned, and therefore repelling each other. By contrast the axial configuration has these dipoles roughly opposing, thus representing a more stable and lower energy state.

Both the hyperconjugation and the dipole minimization contribute to the preferred (Z)-conformation of esters over the (E)-conformation. In the (Z) conformation the lone pair of electrons in the alpha oxygen can donate into the neighboring σ* C-O orbital. In addition, the dipole is minimized in the (Z)-conformation and maximized in the (E)-conformation.[5]

n-n Repulsions and C-H Hydrogen Bonding
If the lone pairs of electrons on the oxygens at the anomeric center of 2-methoxypyran are shown, then a brief examination of the conformations of the anomers reveal that the β-anomer always has at least one pair of eclipsing (coplanar 1,3-interacting) lone pairs, this n-n repulsion is a high energy situation. On the other hand, the α-anomer has conformations in which there are no n-n repulsions, and that is true in the exo-anomeric conformation. The energetically unfavourable n-n repulsion present in the β-anomer, coupled with the energetically favourable C-H hydrogen bond between the axial H-5 and a lone pair of electrons on the axial α-anomeric substituent, have been suggested [references 7 and 8] to account for most of the energetic difference between the anomers, the anomeric effect. The molecular mechanics program StruMM3D, which is not specially parameterized for the anomeric effect, estimates that the dipolar contributions to the anomeric effect (primarily the n-n repulsion, and C-H hydrogen bonding discussed above) are about 1.5 kcal/mol.
Influences on anomeric effect
While the anomeric effect is a general explanation for this type of stabilization for a molecule, the type and amount of stabilization can be affected by the substituents being examined as well as the solvent being studied.'
Substituent effect
In a closed system, there is a difference observed in the anomeric effect for different substituents on a cyclohexane or tetrahydropyran ring (Y=Oxygen). When X=OH, the generic anomeric effect can be seen, as previously explained. When X=CN, the same results are seen, where the equatorial position is preferred on the cyclohexane ring, but the axial position is preferred on the tetrahydropyran ring. This is consistent with the anomeric effect stabilization. When X=F, the anomeric effect is in fact observed for both rings. However, when X=NH2, no anomeric effect stabilization is observed and both systems prefer the equatorial position. This is attributed to both sterics and an effect called the reverse anomeric effect (see below).[3]
Solvent effect
One common criticism of the hyperconjugation theory is that it fails to explain why the anomeric effect is not observed when substituted tetrahydropyran molecules are placed in polar solvents, and the equatorial position is once again preferred. It has been shown, however, that hyperconjugation does depend on the solvent in the system. Each of the substituted systems described above were tested in the gas phase (i.e. with no solvent) and in aqueous solution (i.e. polar solvent). When X=F, the anomeric effect was observed in both media, and the axial position was always preferred. This is attributed to hyperconjugation. When X=OH or CN, the anomeric effect was seen in the gas phase, when the axial position was preferred. However, in aqueous solutions, both substituents preferred the equatorial position. This is attributed to the fact that there are more electrostatic repulsions with the axial positioned substituent and the polar solvent, causing the equatorial position to be preferred. When X=NH2, again, no anomeric effect was observed and the equatorial position was always preferred.[10]
Overcoming the anomeric effect
While the anomeric effect can cause stabilization of molecules, it does have a magnitude to its stabilization, and this value can be overcome by other, more destabilizing effects in some cases.

In the example of spiroketals, the orientation on the upper left shows stabilization by the hyperconjugative anomeric effect twice, thus greatly stabilizing the orientation of the molecule. The orientation on the upper right only shows this hyperconjugative anomeric stabilization once, causing it to be the lesser preferred structure. However, when substituent are added onto the spiroketal backbone, the more preferred structure can be changed. When a large substituent is added to the spiroketal backbone, as seen in the lower left, the strain from having this large substituent, R, in the axial position is greatly destabilizing to the molecule. In the molecule on the lower right, R is now in the equatorial position, which no longer causes destabilization on the molecule. Therefore, without substituents, the upper equilibrium reaction is favored on the left hand side, while the lower equilibrium is favored on the right hand side, simply from the addition of a large, destabilizing substituent.[11]
Exo Anomeric Effect
An extension of the anomeric effect, the exo anomeric effect is the preference of substituents coming off a ring to adopt the gauche conformation, while sterics would suggest an antiperiplanar conformation would be preferred.
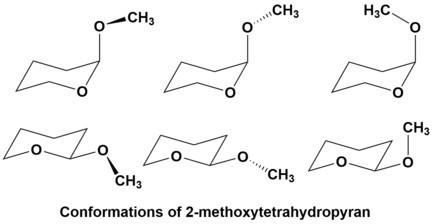
An example of this is 2-methoxytetrahydropyran. As the anomeric effect predicts, the methoxy substituent shows an increased preference for the axial conformation. However, there is actually more than one possible axial conformation due to rotation about the C-O bond between the methoxy substituent and the ring. When one applies the principles of the reverse anomeric effect, it can be predicted that the gauche conformer is preferred, suggesting the top left conformation is best in the figure above. This prediction is supported by experimental evidence. Furthermore, this preference for the gauche position is still seen in the equatorial conformation.[12]
Reverse Anomeric Effect
This term refers to the apparent preference of positively charged nitrogen substituents for the equatorial conformation beyond what normal steric interactions would predict in rings containing an electronegative atom, such as oxygen. Substituents containing carbons with partial positive charges are not seen to exhibit the same effect.[13] There is some debate as to whether or not this is a real phenomenon. The nitrogen containing substituents it has been reported with are quite bulky, making it hard to separate the normal effects of steric bulk and the reverse anomeric effect, if it does exist.[14] For example, in the molecule shown below, the pyridinium substituent strongly prefers the equatorial position, as steric factors would predict, but actually shows a stronger preference for this conformation than predicted, suggesting the reverse anomeric effect is contributing.

Synthetic Applications
The anomeric effect is taken into consideration synthetically. Due to its discovery in sugars, sugar and carbohydrate chemistry is one of the more common synthetic uses of the anomeric effect. For instance, the Koenigs-Knorr Glycosidation installs an α-OR or β-OR group in high diastereoselectivity which is effected by the anomeric effect. Sophorolipid Lactone, (+)-Lepicidin A, and (−)-Lithospermoside are a few of the products synthesized via the Koenigs-Knorr Glycosidation overcoming the anomeric effect.[15]

See also
- Cyclohexane conformation
- Carbohydrate conformation
- Anomer
- Monosaccharide
- Alkane stereochemistry
- Gauche effect
- Steric effects
- Intramolecular forces
- Conformational isomerism
- Raymond Lemieux
References
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (1996) "Anomeric Effect".
- ↑ Juaristi, E.; Cuevas, G. (1992). "Recent studies of the anomeric effect". Tetrahedron. 48 (24): 5019–5087. doi:10.1016/S0040-4020(01)90118-8.
- 1 2 Bauerfeldt, Glauco F.; Cardozo, Thiago M.; Pereira, Márcio S.; da Silva, Clarissa O. (1 January 2013). "The anomeric effect: the dominance of exchange effects in closed-shell systems". Organic & Biomolecular Chemistry. 11 (2): 299. doi:10.1039/c2ob26818c.
- ↑ Kirby, Anthony J. (1983). The anomeric effect and related stereoelectronic effects at oxygen ; with 24 tables. Berlin [u.a.]: Springer. ISBN 0-387-11684-2.
- 1 2 Cuevas, Eusebio Juaristi, Gabriel (1995). The anomeric effect. Boca Raton: CRC Press. ISBN 0-8493-8941-0.
- ↑ Vila, A.; Mosquera, R. A. (2007). "Atoms in molecules interpretation of the anomeric effect in the O—C—O unit". J. Comp. Chem. 28 (9): 1516–1530. doi:10.1002/jcc.20585. PMID 17330885.
- ↑ Box, V. G. S. (1990). "The role of lone pair interactions in the chemistry of the monosaccharides. The anomeric effect". Heterocycles. 31 (6): 1157–1181. doi:10.3987/REV-90-414.
- ↑ Box, V. G. S. (1991). "The role of lone pair interactions in the chemistry of the monosaccharides. Stereo-electronic effects in unsaturated monosaccharides". Heterocycles. 32 (4): 795–807. doi:10.3987/REV-91-425.
- ↑ Sundberg, Francis A. Carey ; Richard J. (2007). Advanced Organic Chemistry : Part A: Structure and Mechanisms (5., ed.). Berlin: Springer US. ISBN 978-0-387-68346-1.
- ↑ Freitas, Matheus P. (1 January 2013). "The anomeric effect on the basis of natural bond orbital analysis". Organic & Biomolecular Chemistry. 11 (17): 2885. doi:10.1039/c3ob40187a.
- ↑ Perron, Francoise; Albizati, Kim F. (1 November 1989). "Chemistry of spiroketals". Chemical Reviews. 89 (7): 1617–1661. doi:10.1021/cr00097a015.
- ↑ Szarek, Walter A. (1979). Anomeric Effect: Origins and Consequences. Washington: American Chemical Society. ISBN 0-8412-0470-5.
- ↑ Kirby, A.J. (1983). The Anomeric Effect and Related Stereoelectronic Effects at Oxygen. New York: Springer-Verlag. ISBN 0-387-11684-2.
- ↑ Thatcher, Gregory R. J. (1993). The Anomeric Effect and Associated Stereoelectronic Effects. Washington: American Chemical Society. ISBN 0-8412-2729-2.
- ↑ Kürti, László; Czakó, Barbara (2007). Strategic applications of named reactions in organic synthesis : background and detailed mechanisms ; 250 named reactions (Pbk. ed., [Nachdr.]. ed.). Amsterdam [u.a.]: Elsevier Academic Press. ISBN 978-0-12-429785-2.